Article Text

Statistics from Altmetric.com
Heart failure with preserved ejection fraction (HFpEF) is estimated to account for about 1/2 of all patients with heart failure. Yet effective medical therapy for HFpEF has been elusive with only minimal benefit from pharmacologic approaches of proven benefit for heart failure with reduced ejection fraction (HFrEF). As the word cardiovascular implies, normal circulatory function depends on normal function of both the heart and the vascular system. Systemic vascular dysfunction is recognized as a key contributor to symptoms and poor outcomes in HFrEF and is a key therapeutic target. However, the role of vascular dysfunction in HFpEF has been less well characterized.
In this issue of Heart, Lee and colleagues (see page 278) provide detailed studies of vascular function in 24 patients with HFpEF compared to 23 control subjects. The function of larger conduit arteries was measured by brachial artery flow mediated dilation whereas microvascular function was assessed by reactive hyperemia after a period of cuff occlusion. They found that flow mediated dilation was no different in patients with HFpEF compared to controls, when normalized for shear stress stimulus. In contrast, microvascular function was abnormal with an approximately 30% reduction in reactive hyperemia in HRpEF patients (figure 1). The authors suggest that the differing types of vascular dysfunction in heart failure – abnormal conduit function in HFrEF versus abnormal microvascular function in HFpEF—might be account for some of the differences in therapeutic responses in these two types of heart failure.

Postocclusion reactive hyperaemia, expressed as absolute blood flow (panel A) and as blood flow area under the curve (AUC; panel B) in patients with heart failure with preserved ejection fraction (HFpEF) and healthy individuals (Controls). Data are presented as mean±SE. *Significantly different from Controls, p≤0.02.
In the accompanying editorial (see page 257), Lam and Lund point out that this data adds support to the emerging view that HFpEF is characterized by primary vascular endothelial cell dysfunction, specifically with coronary artery microvascular changes leading to increased cardiomyocyte stiffness, left ventricular (LV) concentric remodeling and abnormal diastolic function (figure 2). Thus, therapies that directly target endothelial dysfunction, rather than focusing on neuroendocrine activation, may be effective for treatment of HFpEF.

HFpEF paradigm In HFpEF, comorbidities (such as hypertension, overweight, diabetes, chronic kidney disease, chronic obstructive pulmonary disease, anemia and iron deficiency) lead to microvascular inflammation and endothelial activation. This adversely affects the adjacent cardiomyocyte through decreased nitric oxide (NO) bioavailability, reduced cyclic guanosine monophosphate (cGMP) availability, and altered phosphorylation of titin; microvascular ischemia, concentric left ventricular (LV) remodeling and fibrosis from endothelial-mesenchymal transition (EndMT) contributes further to LV diastolic dysfunction. In contrast, in HFrEF, direct cardiomyocyte injury (e.g. acute myocardial infarction, infections, toxins) leads to cardiomyocyte necrosis, cellular apoptosis and eccentric LV remodeling which set up a vicious cycle of compensatory but maladaptive neuroendocrine activation. Figure adapted from Paulus WJ, Tschope C. J Am Coll Cardiol 2013;62:263–71.
A more detailed discussion of the role of oxidative stress and endothelial dysfunction in HFpEF is provided in the Education in Heart article by Franssen and colleagues. (see page 320) This article discusses the association between common clinical conditions such as diabetes, hypertension and obesity and HFpEF, which is characterized by predominant LV diastolic dysfunction. Current data suggests that a low grade inflammatory state leads to oxidative stress and microvascular inflammation, resulting in decreased nitric oxide bioavailability. The downstream effects of reduced nitric oxide, mediated by reduced cyclic guanosine monophosphate (cGMP) activity, include titin phosphorylation with post translational modifications leading to increased cardiomyocyte stiffness (figure 3). This underlying pathophysiology might be amenable to medical therapy directed at increasing levels of cGMP, reducing oxidation stress or suppressing inflammation, as well as optimal control of the co-morbidities that lead to HFpEF.

cGMP-PKG pathway modulation. In the normal situation, NO is generated from L-arginine by eNOS. NO stimulates sGC to catalize the formation of cGMP from GTP. However, cGMP can also be generated by stimulation of pGC by NPs. cGMP exerts its effects via PKG and both have diverse beneficial cardiovascular effects, as indicated in green. In the situation of inflammation and/or increased oxidative stress, NO reacts rapidly with O2−, to form the toxic ONOO- with many deleterious cardiovascular effects, inidicated in red. Moreover, inflammation and oxidative stress uncouple the NO producing eNOS dimer into O2−,generating monomers, further exacerbating oxidative stress. Another effect of oxidative stress and ONOO− is oxidation of sGC, which leads to loss of its heme-group, making sGC dysfunctional and unresponsive to NO and blocking cGMP formation. Diverse therapeutic strategies are shown in blue. Nitroxyl (HNO, a reduced form of NO) is better tolerated and more stable than nitrates and stimulates sGC. pGC can be stimulated by increasing the availability of NPs, which can be achieved with the combined neprilysin/angiontensin receptor blocker LCZ696. sGC in its inactive, NO resistant heme-free form can be targeted by cinaciguat, a sGC activator. Riociguat and vericiguat mimick NO and are able to directly stimulate sGC. Finally, cGMP can be inbitited by blocking PDEs, such as PDE5 and the (probably) myocardial specific PDE9. Abbreviations: NO, nitric oxide; eNOS, endothelial NO synthase; sGC, soluble guanylate cyclase; cGMP, cyclic guanosine 3′,5'-monophosphate; GTP, guanosine 5'-triphosphate; pGC, particulate GC; NP, natriuretic petptide; PKG, protein kinase G; O2 -, superoxide; ONOO-, peroxynitrite; PDE, phosphodiesterase. Adapted and modified with permission from Hobbs AJ, Stasch JP. Soluble Guanylate Cyclase. Allosteric Activation and Redox Regulation. In: Nitric Oxide: Biology and Pathophysiology. Elsevier 2010. 301–26.
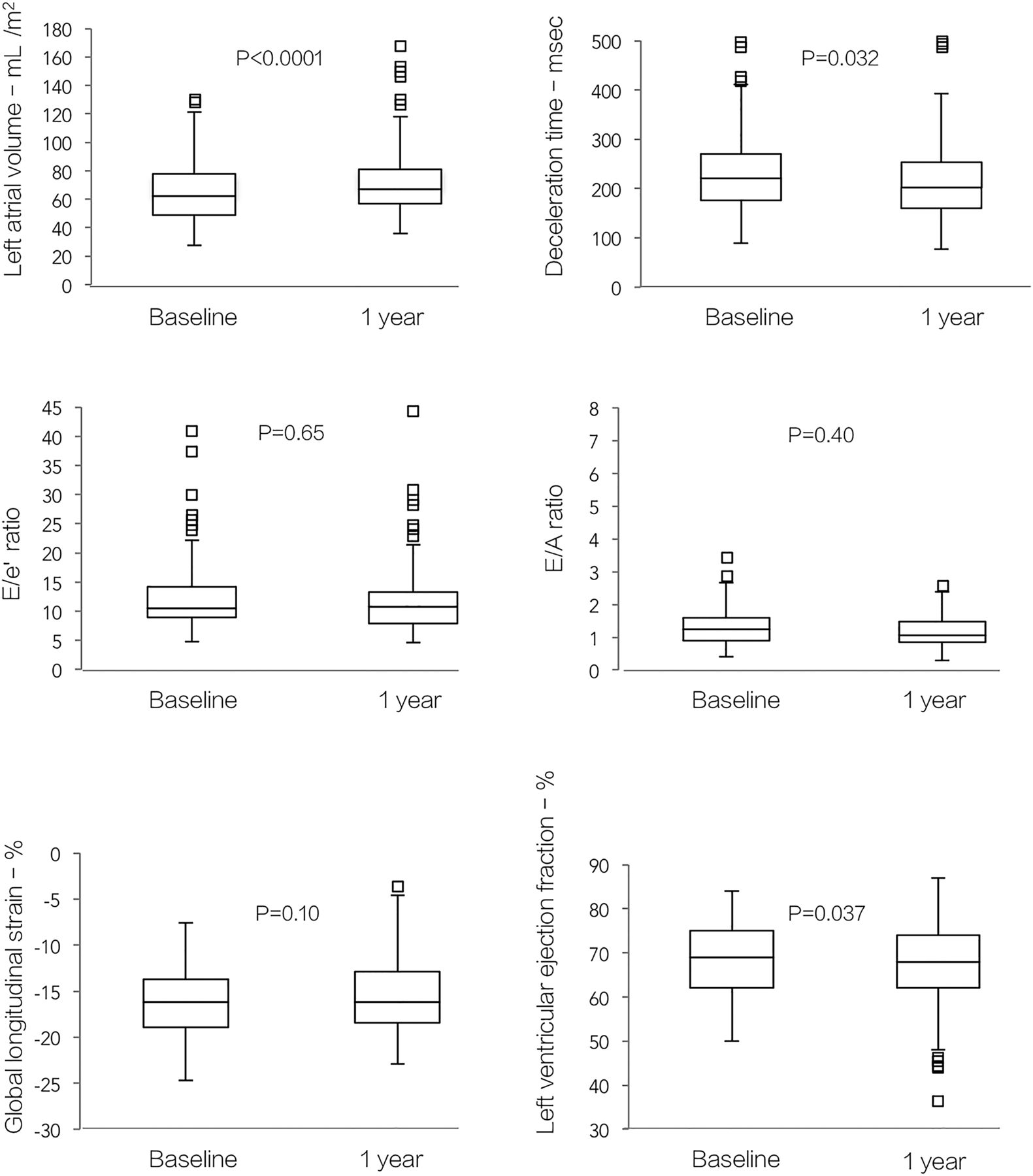
Hypertrophic cardiomyopathy (HCM) is another type of myocardial disease that has few effective medical treatments, with current therapy directed at symptom relief and prevention of complications rather than modification of the disease process itself. Inhibition of the renin-angiotensin system with an angiotensin II receptor blockers (ARB) has been shown to improve LV geometry and function, as well as functional capacity in small pilot studies. To test whether ARB therapy can prevent progressive cardiac dysfunction and improve exercise capacity in a larger clinical population, Axelsson and colleagues (see page 285) initiated the INHibition of the renin angiotensin system in hypertrophic cardiomyopathy and the Effect on hypertrophy Randomized Intervention Trial (INHERIT) study which randomized 133 HCM patients to losartan therapy compared to placebo. As previously reported there was no difference between groups in the primary endpoint of a reduction in LV mass after 12 months. In this issue of Heart, the secondary endpoints of the INHERIT study are presented. Disappointingly, after 12 months of therapy with either losartan or placebo, there also were no differences between groups in LV systolic function, diastolic function, or exercise capacity. In both groups, the disease continued to progress with decline in ejection fraction (mean change −2%, 95% CI −3% to −1%), p=0.037 and an increase in left atrial volume of 6 mL/m2 (95% CI 3 to 9 mL/m2, p<0.0001) (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes in cardiac structure and function from baseline to 1-year follow-up for both groups (n=133). The results did not differ with treatment group. p Values are for changes from baseline to follow-up.
In an editorial, Dhillon and Desai comment (see page 260) that this data supports the concept that LV hypertrophy in HCM and hypertensive heart disease are due to different pathophysiologic processes, with ARB therapy being effective in reversing LV hypertrophy in hypertensive heart disease but not in HCM. However, we also need to consider the possibilities that ARB therapy needs to be started much earlier in the disease course or that follow-up at 1 year is not long enough for effects to be evident. They conclude: “We are entering an era where there is an emerging necessity to develop guidelines for HCM (pharmacotherapies, invasive therapies and risk stratification recommendations) based on sound scientific, and preferably prospective data, rather than just “expert” consensus opinions. Axelsson et al have taken one of the first steps in this direction.”
The Image Challenge case in this issue (see page 277) shows both echocardiographic images and coronary angiography with the question asking for the cause of the patient's chest pain. Several online videos enhance the learning experience.
Linked Articles
- Education in Heart
- Heart failure and cardiomyopathies
- Heart failure and cardiomyopathies
- Editorial
- Review
- Editorial