Article Text

Abstract
Amyloid light chain (AL) amyloidosis is a systemic disease characterised by the aggregation of misfolded immunoglobulin light chain (LC), predominantly in the heart and kidneys, causing organ failure. If untreated, the median survival of patients with cardiac AL amyloidosis is 6 months from the onset of heart failure. Protracted time to establish a diagnosis, often lasting >1 year, is a frequent factor in poor treatment outcomes. Cardiologists, to whom patients are often referred, frequently miss the opportunity to diagnose cardiac AL amyloidosis. Nearly all typical cardiac support measures, with the exception of diuretics, are ineffective and may even worsen clinical symptoms, emphasising the need for accurate diagnosis. Patients with severe cardiac involvement face poor outcomes; heart transplantation is rarely an option because of multiorgan involvement, rapid clinical decline and challenges in predicting which patients will respond to treatment of the underlying plasma cell disorder. Early diagnosis and prompt treatment with ‘source therapies’ that limit the production of amyloidogenic LC are associated with better survival and improvement in organ function after a median of 2.4 months following haematological complete response. However, organ recovery is often incomplete because these source therapies do not directly target deposited amyloid. Emerging amyloid-directed therapies may attenuate, and potentially reverse, organ dysfunction by clearing existing amyloid and inhibiting fibril formation of circulating aggregates. Improved recognition of AL amyloidosis by cardiologists allows for earlier treatment and improved outcomes.
- Cardiac imaging and diagnostics
- Infiltrative cardiomyopathies
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
In amyloid light chain (AL) amyloidosis, misfolded immunoglobulin light chain (LC) produced by clonal plasma cells deposits in organs, causing their dysfunction (figure 1). The disease predominantly (~70% of patients) affects the heart, kidneys or both.1 2 Advanced (Mayo stages IIIa, IIIb) cardiac AL amyloidosis is particularly lethal; median survival is <1 year versus ~8 years in patients without cardiac involvement.2
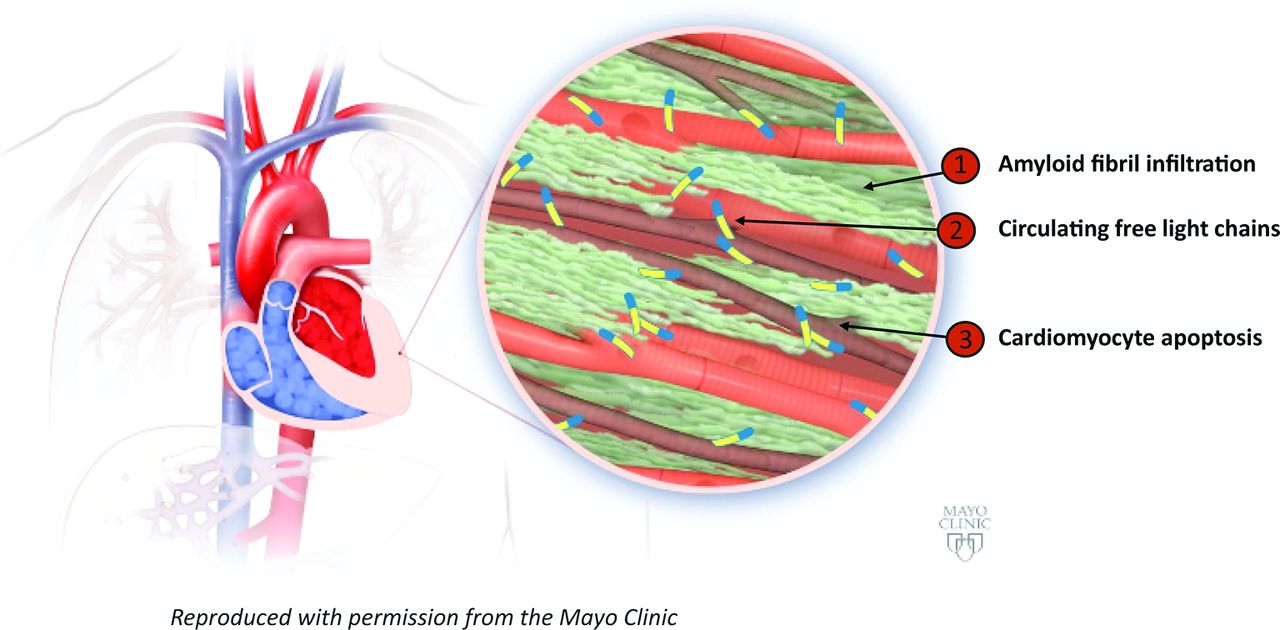
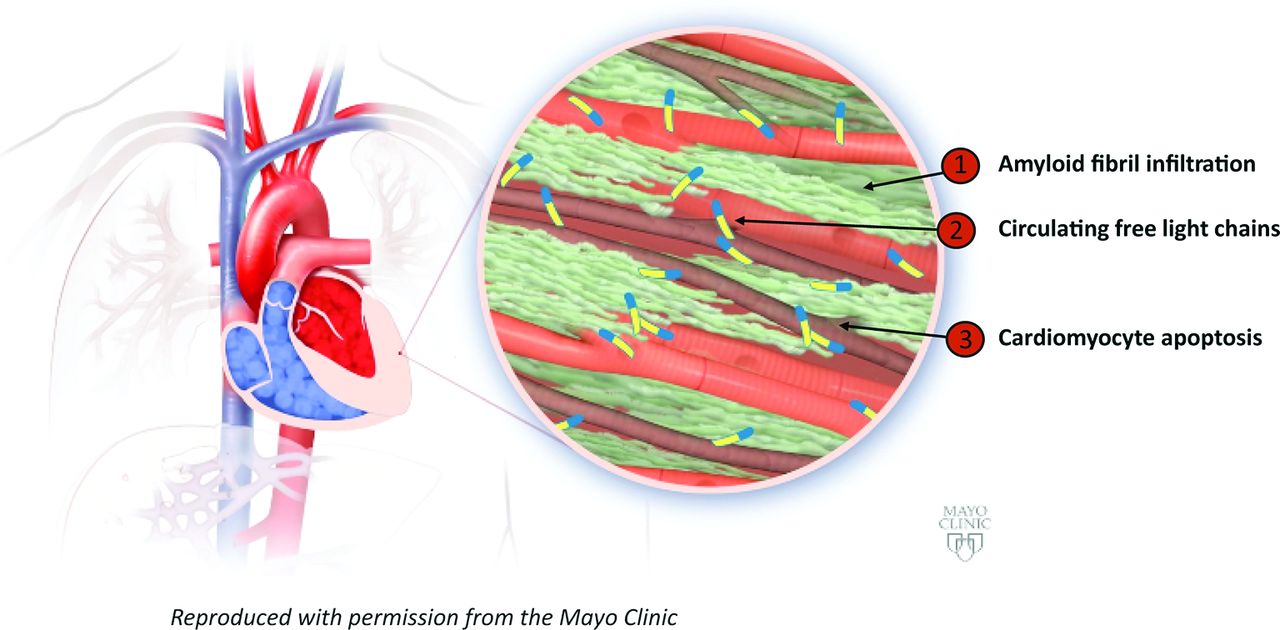
Pathophysiology of amyloid light chain (AL) amyloidosis illustrating three key mechanisms: (1) amyloid fibril infiltration, depicted as green, results in wall thickening and diastolic dysfunction. (2) Local effects of fibril infiltration contribute to myocyte dysfunction and apoptosis. (3) Circulating free light chains contribute to myocardial dysfunction. Reproduced with permission from the Mayo Clinic.
In AL amyloidosis, LC fibrils infiltrate the myocardium, interfere with cell–cell coupling, disrupt cellular integrity3 4 and may contribute to cell injury and death. LC monomers also can elicit oxidative damage by interacting with proteins involved in cell viability and metabolism,5 suggesting that LC may be internalised. LC may promote cardiac stress hormone brain natriuretic peptide (BNP) expression through p38 mitogen-activated protein kinase (MAPK).6 Sudden death in patients with AL amyloidosis is usually attributed to pulseless electrical activity, followed by ventricular arrhythmias, but may result from thromboembolic complications or bradyarrhythmias and conduction system disease secondary to amyloid infiltration or autonomic dysfunction.7
Systemic amyloidoses, including AL amyloidosis, are frequently undiagnosed.8 Despite advances in therapeutic regimens to stanch AL amyloidosis LC production—so-called source therapies—and to increase overall survival, the frequency of sudden death within ≤90 days of diagnosis remains at 25%–30%7 and likely reflects advanced organ dysfunction. Thus, a critical need exists for earlier diagnosis of AL amyloidosis by cardiologists, to whom most patients without diagnoses are initially referred but who, based on a recent patient survey, diagnose amyloidosis in only ~18% of patients.8 The other primary cause of cardiac amyloidosis is deposition of mutant or wild-type transthyretin amyloid (ATTRm or ATTRwt amyloidosis, respectively), which is outside the focus of this review but is similarly important (table 1).
Comparison of causes, clinical presentation and clinical outcome of the main types of cardiac amyloidosis
Cardiologist identification of patients with cardiac AL amyloidosis
AL amyloidosis may develop in patients with multiple myeloma (10%–15%) or may progress from monoclonal gammopathy of undetermined significance (MGUS) (9%).9 These disorders are thus important diagnostic considerations; however, AL amyloidosis can be diagnosed in the absence of either disorder. The overall incidence of AL amyloidosis is estimated to be only 8.0–14.4 million persons per year in the USA10 and suggests that a physician may diagnose only a few cases annually. Nonetheless, based on its prevalence, most cardiologists have encountered patients with cardiac AL amyloidosis. The nature of the patients’ cardiac dysfunction may go undiagnosed, however, because symptoms of cardiac AL amyloidosis may mimic those of other restrictive cardiomyopathies, despite their being predominantly caused by AL amyloidosis (see online supplementary table 1).
Online supplementary table 1
Signs and Symptoms of Cardiac Amyloidosis.
AL amyloidosis is rarely diagnosed before symptoms appear; the vague, variable signs and symptoms of AL amyloidosis reflect advanced organ manifestation or involvement. Clues to diagnosis include heart failure associated with neuropathy, nephrotic syndrome, hepatomegaly, periorbital bleeding and macroglossia. Cardiac ATTR amyloidosis is also frequently associated with neuropathy, primarily in patients with ATTRm amyloidosis, again highlighting the need for physician awareness in differential diagnosis. For patients with many forms of systemic amyloidosis, diagnosis ≥1 year after initial presentation to a physician is, unfortunately, common. It is estimated that one-third of patients may visit five or more physicians before diagnosis.8 Early detection allows for treatments that can halt cardiac damage, potentially reducing the risk for sudden death7 and allowing for spontaneous or therapeutically induced amyloid clearance and improved cardiac function.
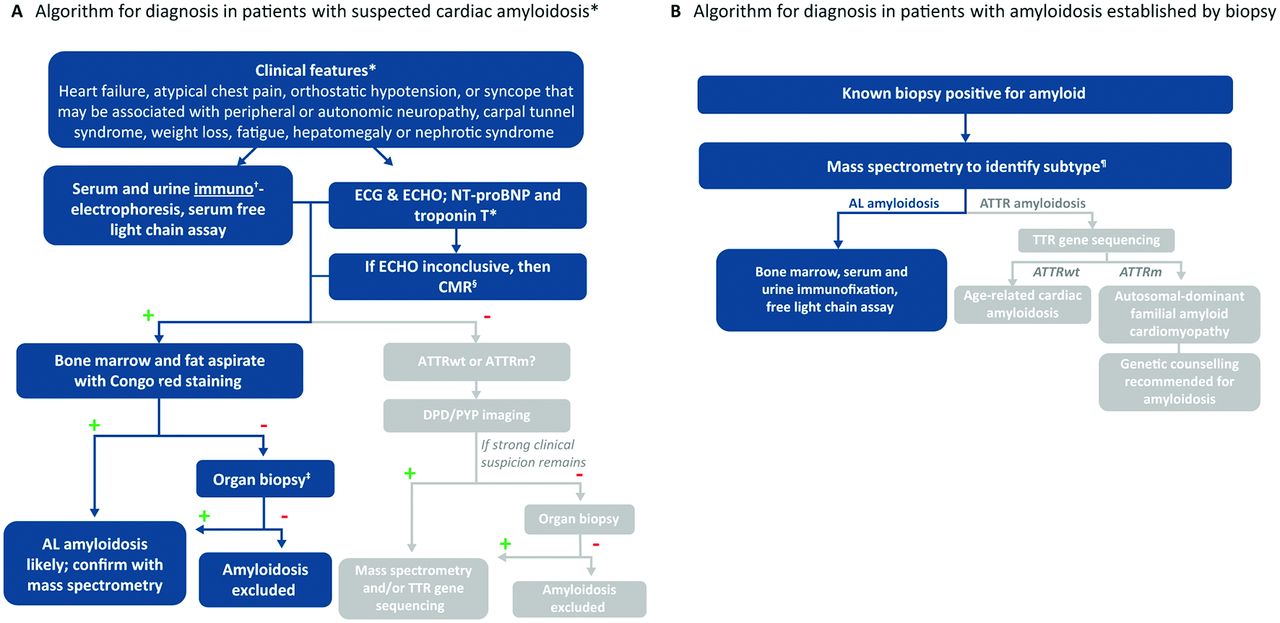
The diagnosis of suspected AL amyloidosis, irrespective of organ involvement, involves identification of clonal disease and amyloid typing (figure 2). Serum-free light chain (FLC) should be measured, and serum and urine immunoelectrophoresis should be performed in all patients. If indicated based on initial screening tests, bone marrow biopsy is performed to determine whether plasma cell dyscrasia is present and to determine the percentage and type of λ- or κ-producing plasma cells.11 Routine use of the immunoglobulin FLC assay in patients with unexplained heart failure may be a relatively efficient, economical and non-invasive means to screen patients with AL amyloidosis. Nonetheless, healthcare providers must recognise that ATTR commonly affects the heart and will not be detected by the serum FLC assay. ATTRwt is an increasingly recognised form of amyloid, particularly in men older than 60 years.

Diagnostic algorithm for cardiac amyloidosis. In patients with diagnosed amyloid light chain (AL) amyloidosis, NT-proBNP and troponin measures carry prognostic significance. *Clinical presentation does not distinguish AL from ATTR amyloidosis. †Serum protein electrophoresis without immunofixation is not sufficient. ‡Heart or other affected organ, proceeding with organ biopsy along either arm of the algorithm, combined with mass spectrometry when positive, allows for the identification of rare types of cardiac amyloid. §Depending on local expertise, CMR can be considered as an initial imaging test. ¶Although mass spectrometry is available at only a few centres, specimens can be sent for review; immunohistochemical techniques may lead to inaccurate typing. +, positive test result; –, negative test result; AL, amyloid light chain amyloidosis; ATTRm, hereditary transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis; CMR, cardiac MR; DPD, 99m3,3diphosphono-1, 2-propanodicarboxylic acid; ECHO, echocardiography; NT-proBNP, N-terminal fragment of pro-brain natriuretic peptide; PYP, 99mTc-pyrophosphate. Modified with permission from Gertz M et al, Nat Rev Cardiol 2015;12:91–102.
Diagnosis must be confirmed by the detection of amyloid in organ or other tissue biopsy, using Congo red or other histological staining, and accurate typing. The choice of biopsy site depends on clinical findings and local expertise. Cardiac biopsy conclusively identifies cardiac AL amyloidosis; however, initial assessment of amyloid from periumbilical fat aspirates or from bone marrow or labial salivary gland biopsy specimens is a less invasive approach in patients with suspected AL amyloidosis with relatively high sensitivity (~80%) treated in experienced centres. Accurate amyloid typing is critical because treatment of cardiac amyloidosis depends entirely on amyloid type. Inaccurate typing exposes patients to inappropriate and ineffective treatment regimens with significant treatment-related morbidity and mortality. The current gold standard of amyloid typing is to determine the precursor protein using laser microdissection mass spectrometry. Although, this is available in only a few centres, specimens—including those fixed in paraffin—can be sent for analysis. Immunohistochemistry techniques have been misleading, even in experienced centres,12 13 and are discouraged. Mass spectrometry amyloid typing is not limited by selection of specific antibodies, thus allowing proper identification of the rarer forms of amyloid (eg, AA, apolipoprotein A1, gelsolin) that may affect the heart.
Organ manifestation or involvement can be assessed using a combination of biomarker assays, imaging assessments and functional tests (table 2). Cardiac involvement is defined by consensus guidelines as mean left ventricular (LV) wall thickness ≥12 mm in the absence of other causes, N-terminal fragment of the pro-brain natriuretic peptide (NT-proBNP) >332 pg/mL in the absence of renal failure or atrial fibrillation or both. Cardiac biomarkers are the most robust predictors of prognosis in AL amyloidosis14 15 and underlie the Mayo 2004 and 2012 staging systems.16 17 Of note, the general features of cardiac involvement detailed in the aforementioned criteria and the functional assessments enumerated below, and in figure 3 and online supplementary table 1, do not specifically define cardiac AL amyloidosis in many patients because the presentation of disease is heterogeneous, but they do define the most common cardiac AL amyloidosis features. Furthermore, the simultaneous interpretation of a patient’s ECG, echocardiographic and imaging data can be instrumental in evaluating the relationship between functional and structural cardiac changes.

Examples of patient assessments showing cardiac involvement. (A) ECG of a 62-year-old patient with amyloid light chain (AL) amyloidosis initially diagnosed with hypertrophic cardiomyopathy. (B) Although the limb lead voltage is slightly above the threshold for low voltage, the voltage is discordant with the degree of left ventricular wall thickening (interventricular septum and posterior wall both measure 18 mm). ECG demonstrated an anteroseptal infarct pattern, but no regional wall motion abnormalities were observed and the patient had no history of myocardial infarction. (C) The typical echocardiographic pattern of abnormal global longitudinal strain in cardiac amyloid is demonstrated with markedly abnormal basal function and preserved apical function leading to a bull's eye pattern. This pattern may be a clue to cardiac amyloid when other echocardiographic findings are subtle or absent. (D) Cardiac MRI in amyloidosis demonstrating difficulty ‘nulling’ the myocardium (myocardium appears similar to blood pool) and diffuse abnormalities of delayed gadolinium enhancement. CMR, cardiovascular MR; ECHO, echocardiography.
Consensus criteria for organ involvement
Patients with MGUS are at increased risk for AL amyloidosis. The differentiation of AL amyloidosis is based on the detection of organ damage and associated LC amyloid. Tissue diagnosis is important given that MGUS is common in patients with ATTRwt, but it is unrelated to amyloid deposition in these patients.
Electrocardiography
Infiltration of cardiac tissue with LC deposits is associated with electrical disturbances.3 Several studies have observed lowered QRS voltage (≤5 mV), particularly evident in limb leads, with cardiac AL amyloidosis.17 18 This finding distinguishes infiltrative from hypertrophic cardiac conditions and may be apparent at early stages of cardiac involvement3 and correlated with worse outcome.19 Low-voltage QRS complex occurs in approximately 45% of patients with AL amyloidosis and is more common than in patients with ATTR amyloidosis, perhaps related to the direct myocardial toxic effects of circulating LC independent of the degree of infiltration.17 Pseudo-infarct patterns are present in approximately 45% of patients with AL amyloidosis18 and should prompt consideration of amyloid in patients with ECG infarct patterns in the absence of regional wall motion abnormalities. Fragmented QRS is a common finding in AL amyloidosis and is associated with worse prognosis.20 The absence of low-voltage QRS complex does not exclude the diagnosis of AL amyloidosis; in fact, in one series, 16% of patients with cardiac biopsy-proven AL amyloidosis met ECG criteria for LV hypertrophy.18 Simultaneous interpretation of ECG and echocardiographic findings, as discussed below, is an important strategy.
Echocardiography
Increased cardiac wall thickness without a known cause (eg, uncontrolled hypertension) may be the first red flag for cardiac amyloidosis. Characteristic echocardiographic findings in amyloidosis include increased left and right ventricular wall thickness with greater echogenicity (granular sparkling), though the latter is less specific in the era of harmonic imaging, and normal or mildly reduced LV cavity size with enlarged atria.17 21Patients typically have preserved or moderately decreased LV ejection fraction (LVEF) but poor longitudinal function, possibly an early sign of cardiac AL amyloidosis. A characteristic LV strain pattern with preservation of the apex (bull's eye) is often an indication of the disease. Diastolic dysfunction is frequently present, usually with a restrictive pattern (grade II-IV diastolic dysfunction) consistent with increased LV filling pressure.3 Normal or grade I diastolic dysfunction is rare in AL amyloidosis with significant cardiac involvement. Echocardiography often shows less severe wall thickening in AL cardiomyopathy than in ATTR amyloidosis, despite more severe heart failure and a more aggressive clinical course.17 Abnormalities of longitudinal ventricular function demonstrated by strain imaging are independent predictors of survival.22 Furthermore, abnormal right ventricular strain may be an early diagnostic clue.23 The severity of echocardiographic abnormalities and the rapidity at which they develop may correlate with worse prognosis.19 Atrial thrombi are common in AL amyloidosis, detected by echocardiography in 35% of patients in one series, and may occur even in the presence of sinus rhythm.24
Although most patients with significant cardiac AL amyloidosis have increased LV wall thickness, heart failure can occur in those with normal wall thickness.25 26 This may be explained by cardiotoxicity of the circulating LCs causing myocardial dysfunction, which in some patients may predominate over fibril deposition. In addition, patients with wall thickness in the lower normal range at baseline may have significant amyloid deposition, yet the measured wall thickness may fall within the ‘normal’ range for age, sex and body surface area. An important distinction is that patients with ATTR, as opposed to AL, amyloidosis can experience a longer period of chronic disease before diagnosis, which may account for the increased wall thickness.
Although cardiac AL amyloidosis is generally considered a condition in which LVEF is preserved, a subset of patients may present with reduced LVEF and minimal or no ventricular wall thickening, again perhaps because of direct LC toxicity or predominant vascular involvement.26 Last, some patients with AL amyloidosis present with primarily endocardial involvement, leading to atypical imaging findings with thickened valves and endocardial surfaces but normal or only mildly increased wall thickness.27
Cardiac MR (CMR) and nuclear imaging
CMR and nuclear imaging should be considered for patients with unexplained heart failure and arrhythmias, especially when there is a suspicion of amyloid.28 Difficulty nulling the myocardium, abnormality of late gadolinium enhancement and abnormal T1 signal are all characteristic findings of cardiac amyloidosis. CMR is particularly helpful when the echocardiogram is inconclusive, and it is often the first clue to cardiac amyloidosis when the diagnosis has not otherwise been considered. However, CMR does not distinguish amyloid type, is limited because of restrictions in patients with renal impairment19 and pacemakers/defibrillators and does not definitively establish the diagnosis. Furthermore, patchy deposits may not cause abnormalities of late gadolinium enhancement and may lead to false-negative results. Two newer imaging agents, N-[methyl-11C]2-(49-methylamino-phenyl)-6-hydroxybenzothiazole (11C-PIB; 11C-Pittsburgh B compound)29 and florbetapir,30 can specifically bind cardiac amyloid and may be useful when combined with structural and functional imaging assessments. Although these agents are not specific to LC amyloid, florbetapir may offer significantly higher binding to LC than transthyretin amyloid. Imaging with 99mTc-pyrophosphate (PYP) and 99m3,3diphosphono-1, 2-propanodicarboxylic acid (DPD) may be useful for identifying ATTR amyloidosis31 and has led to increased awareness of this condition. In the absence of evidence of plasma cell dyscrasia (abnormal serum or urine electrophoresis and/or abnormal FLC), a strongly positive PYP or DPD scan can establish the diagnosis of ATTR amyloidosis without tissue biopsy.32 If a monoclonal protein is present, tissue confirmation of amyloid type is mandatory because nuclear scintigraphy may be mildly positive in AL amyloidosis.
Biomarkers
Several cardiac hormones are secreted by cardiomyocytes during cardiac stress. As such, these hormones are non-specific indicators of cardiac dysfunction; however, cardiac biomarkers tightly correlate with prognosis and therapeutic response in AL amyloidosis.14 33 Current Mayo staging systems for this disease are based on serum levels of NT-proBNP and cardiac troponin T and the concentration of circulating amyloidogenic FLC.15 16 This staging system assigns patients a score of 1 for each differential free light chain (dFLC) ≥18 mg/dL, cTnT ≥0.025 ng/mL, and NT-proBNP ≥1800 pg/mL, creating stages I (no points) to IV (three points). Median overall survival is greater for patients with lower stage of disease (94.1 months (stage I), 40.3 months (II), 14 months (III) and 5.8 months (IV)). This classification system was validated in the other data sets.16 In clinical practice, physicians may be particularly suspicious of a patient presenting with very high NT-proBNP and troponin levels given a diagnosis of non-obstructive hypertrophic cardiomyopathy. Several other biomarkers, including soluble suppression of tumorigenicity 2,34 growth differentiation factor 15,35 midregional proadrenomedullin36 and osteoprotegerin,37 may prove valuable prognostic indicators.
NT-proBNP is predictive of survival in patients with newly diagnosed disease and in patients who have undergone systemic therapy,14 likely because of the direct mechanism by which LC modulates p38 MAPK, which can directly promote BNP expression.6 Increased cardiac troponin levels indicate disease progression and poor survival in patients with AL amyloidosis, but their role in predicting cardiac response is less established.38 39 Given that delayed diagnosis has a major impact on outcome in patients with cardiac AL amyloidosis, assessment of FLC levels in patients with symptoms of heart failure—and especially in those with elevated NT-proBNP and preserved LVEF—is warranted.
Levels of NT-proBNP are elevated in patients with renal insufficiency and, therefore, may be influenced by reduced glomerular filtration rate and cardiac function in patients with both renal and cardiac amyloidosis. However, although decreasing glomerular filtration rate required a higher threshold for detecting heart involvement and predicting survival, NT-proBNP levels have been found to predict survival in patients with glomerular filtration rates ≥15 mL/min/1.73 m2.40
Treatment of patients with AL amyloidosis
There are no approved therapies for AL amyloidosis; instead, modified treatment regimens for multiple myeloma are generally used. The ultimate goal of therapy is improved organ function; optimal therapies will target both the clonal plasma cell disorder (to stop production of the precursor protein) and existing amyloid deposits in organs and tissue (table 3).
Treatments for patients with cardiac amyloid light chain (AL) amyloidosis and clinical outcome
Plasma cell-directed therapies
Treatment regimens are based primarily on those for multiple myeloma and will reduce amyloidogenic LC precursor protein production from plasma cells; these regimens are the topic of evolving treatment algorithms.41Patients are frequently treated with high-dose chemotherapy in combination with autologous stem cell transplantation (ASCT), alkylating agents, steroids, proteasome inhibitors and/or immunomodulatory drugs. The Mayo Stratification of Myeloma and Risk-Adapted Therapy consensus statement41 recommends initial stratification of patients with newly diagnosed AL amyloidosis to determine their eligibility for ASCT and indicates agents that may offer improved outcomes for first-line and second-line therapy (figure 4). Only a small number of patients with severe cardiac involvement are eligible for ASCT. Nonetheless, patient heterogeneity and the lack of clinical trial data, both heavily influenced by the relative rarity of the condition, confound these guidelines. Overall, plasma cell-directed (source) therapies can reduce the concentration of the proteotoxic LC by ≥50% (partial response or better) in ~60% of patients, which sometimes results in organ response; however, such improvement is often partial or incomplete. Moreover, sudden death can occur even in patients who experience haematological response (HR). In the first-line setting, cardiac response defined by consensus criteria (>30% and>300 pg/mL decrease in NT-proBNP) is observed in ~25% of patients who experience partial HR or better.42–44 In addition, patients who do not achieve organ response to first-line therapy have a lower probability of achieving organ response to second and subsequent lines of therapy. Although cardiac response is often observed over time among patients who achieve HR, particularly those who achieve complete HR,41 treatment to specifically and more rapidly promote organ response is a critical unmet need for all patients. Although cardiac imaging studies are critical in the diagnosis of AL and often provide the first clue to diagnosis, use of these techniques to assess organ response is challenging. Wall thickness measurements are subject to variation based on multiple technical issues, and assessment of diastolic function can be affected by loading conditions and heart rate. Serial assessment of strain has been shown to correlate with clinical response but is subject to variability and differences between imaging platforms. Cardiac MRI findings are subject to similar limitations, though serial T1 mapping holds promise in assessing response to therapy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Suggested plasma cell-directed treatment for patients with newly diagnosed disease based on their Mayo stage, as described in the mSMART guidelines.41 ASCT, autologous stem cell transplantation; BMPC, bone marrow plasma cell; CRAB criteria for myeloma (C = calcium (elevated), R = renal failure, A = anaemia, B = bone lesions); CyBorD, cyclophosphamide, bortezomib, dexamethasone; dFLC, difference in serum free light chain; Mel-Dex, melphalan–dexamethasone; PR, partial haematological response (dFLC decrease ≥50%; assessable in patients with baseline dFLC ≥50 mg/L); VGPR, very good partial haematological response (defined as dFLC <40 mg/L).
Cardiac support therapies
Heart failure caused by cardiac AL amyloidosis poses a unique therapeutic challenge because of the complex nature of cardiac dysfunction in patients with renal and autonomic comorbidities and because typically used supportive measures are contraindicated. Conventional medications, such as beta-blockers, angiotensin-converting enzyme inhibitors and angiotensin-receptor blockers, may contribute to early mortality and worsening of symptoms,7 further underscoring the importance of prompt diagnosis in directing treatment. Diuretics (loop diuretics and aldosterone receptor antagonists), in addition to monitoring electrolytes and creatine, are predominant supportive treatments.7 Use of an LV assist device presents challenges in patients who have small LV cavities with thickened walls and coexisting right ventricular dysfunction.45Implantable cardioverter defibrillators (ICDs) have not been associated with survival benefit in patients with cardiac AL amyloidosis in retrospective series.46 Use of ICDs in selected patients with cardiac AL amyloidosis, especially those meeting the usual criteria for device implantation, remains controversial. Biventricular circulatory support is necessary for most patients, but a total artificial heart is available only as a bridge to transplantation. Concerns regarding infection, bleeding and thrombosis pose additional challenges for the use of circulatory support devices in patients with cardiac AL amyloidosis. As devices evolve, opportunities for circulatory support will likely improve.
Heart transplantation
In addition to the typical constraints associated with organ transplantation, the inadequacy of pretransplantation cardiac support measures and the risk for re-deposition of amyloid in the transplanted heart diminish the success of heart transplantation.47 Nonetheless, some patients with isolated cardiac involvement may be eligible for heart transplantation and may experience enhanced survival. Emerging therapeutic developments may lead to improved organ function, thereby reducing the need for heart transplantation.11
Emerging treatments: amyloid-directed therapies
Immunotherapeutics
Antibody-mediated phagocytosis combined with the clearance of amyloid is a promising approach to reverse organ dysfunction. Three antibodies are in clinical development.
An interim phase 1/2 report of 27 patients with AL amyloidosis treated with NEOD001 (ClinicalTrials.gov, NCT01707264), an antibody that targets misfolded LC, was recently published,48 and confirmatory results in an expansion phase of this study have been presented.49 Favourable renal and cardiac responses were observed in evaluable patients. NEOD001 was well tolerated; no patients experienced infusion-related hypersensitivities. Reductions in NT-proBNP underscore the potential for increasing survival with NEOD001. Moreover, organ responses were evident in patients >2 years after their last plasma cell-directed therapy, supporting that NEOD001 directly improves organ function. Two ongoing randomised, placebo-controlled, global trials (ClinicalTrials.gov, NCT02632786 and NCT02312206) specifically address the safety and efficacy of NEOD001 in patients with cardiac AL amyloidosis.
Richards et al,50 in their interim phase 1 trial report (ClinicalTrials.gov, NCT01777243), observed reduced serum amyloid P (SAP) component, an amyloid-binding protein that stabilises fibrils and promotes fibril formation after a single anti-SAP antibody infusion, in eight patients with AL amyloidosis also treated with the small molecule circulating SAP-binder CPHPC ((2R)−1-[6-[(2R)−2-carboxypyrrolidin-1-yl]−6-oxohexanoyl] pyrrolidine-2-corboxylic acid). This trial detailed two patients with AA (inflammatory) amyloidosis, one patient with AApoA1 amyloidosis and four patients with AFib amyloidosis. Treatment was associated with increased liver resiliency in patients with liver stiffness, a sign of amyloid infiltration. Although the clinical significance of these changes is unknown, this trial validates the immunotherapeutic approach; cardiac responses were not addressed. The safety of anti-SAP treatment, however, requires further study; several patients treated with anti-SAP experienced infusion-related inflammatory responses.
Preliminary results of a phase 1a trial (ClinicalTrials.gov, NCT02245867) of eight patients who previously received systemic treatment and were each administered a single infusion of 11–1 F4, which, like NEOD001, targets misfolded LC, have also been promising. In this small ongoing study, organ responses were observed within 8 weeks after treatment.51
Antifibril formation therapy
Preclinical studies have shown that doxycycline inhibits fibrillogenesis.52 Preliminary results of a case-matched study of 30 patients with stage III cardiac AL amyloidosis (Alchemy; UK National Amyloidosis Centre)53 demonstrated extended survival in response to twice-daily doxycycline (100 mg) and meticulous standard of care. If confirmed, these results may bolster the efficacy of doxycycline in patients who achieve HR.
Conclusion
Delays in diagnosis and treatment likely contribute to the poor life expectancy of patients with cardiac AL amyloidosis despite improved treatments; increased clinical suspicion among cardiologists is needed. Improved survival in patients with less advanced disease highlights that cardiac AL amyloidosis is not a hopeless condition and may be effectively treated. Accurate diagnosis and appropriate treatment will ensure patients are not needlessly subjected to standard medications for heart failure that may be ineffective or even harmful. Emerging interventions may specifically target amyloid, potentially improving cardiac function.
Acknowledgments
Medical editorial assistance was provided by Sara Glickstein, PhD, of ApotheCom (San Francisco, California) and was funded by Prothena Biosciences Inc, whichhad no role in determining content or drafting this article.
References
Footnotes
Contributors All authors wrote and revised the manuscript and approved the final version for submission.
Competing interests MG has received research grants from Pfizer and Alnylam and consulting fees from Prothena Biosciences. AD has received research grants from Celgene, Takeda, Pfizer, Janssen and Alnylam and a travel grant from Roche. MAG has received honoraria from Prothena Biosciences, Amgen, Celgene, Onyx, Novartis and Sandoz.
Provenance and peer review Not commissioned; externally peer reviewed.