Article Text

Abstract
Background The effect of FBN1 mutation type on the severity of cardiovascular manifestations in patients with Marfan syndrome (MFS) has been reported with disparity results.
Objectives This study aims to determine the impact of the FBN1 mutation type on aortic diameters, aortic dilation rates and on cardiovascular events (ie, aortic dissection and cardiovascular mortality).
Methods MFS patients with a pathogenic FBN1 mutation followed at two specialised units were included. FBN1 mutations were classified as being dominant negative (DN; incorporation of non-mutated and mutated fibrillin-1 in the extracellular matrix) or having haploinsufficiency (HI; only incorporation of non-mutated fibrillin-1, thus a decreased amount of fibrillin-1 protein). Aortic diameters and the aortic dilation rate at the level of the aortic root, ascending aorta, arch, descending thoracic aorta and abdominal aorta by echocardiography and clinical endpoints comprising dissection and death were compared between HI and DN patients.
Results Two hundred and ninety patients with MFS were included: 113 (39%) with an HI-FBN1 mutation and 177 (61%) with a DN-FBN1. At baseline, patients with HI-FBN1 had a larger aortic root diameter than patients with DN-FBN1 (HI: 39.3±7.2 mm vs DN: 37.3±6.8 mm, p=0.022), with no differences in age or body surface area. After a mean follow-up of 4.9±2.0 years, aortic root and ascending dilation rates were increased in patients with HI-FBN1 (HI: 0.57±0.8 vs DN: 0.28±0.5 mm/year, p=0.004 and HI: 0.59±0.9 vs DN: 0.30±0.7 mm/year, p=0.032, respectively). Furthermore, patients with HI-FBN1 tended to be at increased risk for the combined endpoint of dissection and death compared with patients with DN-FBN1 (HR: 3.3, 95% CI 1.0 to 11.4, p=0.060).
Conclusions Patients with an HI mutation had a more severely affected aortic phenotype, with larger aortic root diameters and a more rapid dilation rate, and tended to have an increased risk of death and dissections compared with patients with a DN mutation.
- Marfan syndrome
- aortic dilation
- aortic dissection
- FBN1
- haploinsufficiency
Statistics from Altmetric.com
Introduction
Marfan syndrome (MFS) is a connective tissue disorder caused by mutations in the FBN1 gene (NM_000138.4) encoding the fibrillin-1 protein.1 Patients with MFS often have aortic dilation, requiring aortic surgery to prevent aortic dissection.2 However, phenotypic expression and age at manifestations onset are known to vary widely in patients with MFS.3 Over 2900 different FBN1 mutations have been described, which can be classified as dominant negative (DN) or haploinsufficiency (HI).3–8 DN mutations lead to disturbed folding of the protein owing to interference of the mutated with the non-mutated fibrillin-1 protein, resulting in a disorganised extracellular matrix.9 On the other hand, HI mutations lead to production of the only non-mutated fibrillin-1 protein.10–12 Efficient fibrillin-1 deposition into the matrix is reliant on a critical threshold of normal fibrillin-1 accumulation at the cell surface, and the loss of contribution from one allele is sufficient to significantly impair efficient use of protein derived from non-mutated allele. However, matrix incorporation of mutant fibrillin-1 (unique to DN-type mutations) can also promote proteolytic clearance of microfibrils over time, perhaps further exacerbating the functional deficit and contributing to phenotypic progression.13 We recently found that patients with an HI-FBN1 mutation more often have aortic dissections and shorter survival compared with those with a DN-FBN1 mutation.14
The aim of this study was to assess the impact of FBN1 mutation (HI vs DN) on the aortic dilation rate and cardiovascular events in a well-controlled MFS cohort.
Methods
Patient population
All children and adults diagnosed with MFS, carrying a pathogenic FBN1 mutation and who were followed up at two specialised MFS centres in Spain between 2004 and 2015 were included. The Ethics committee of Hospital Vall d’Hebron approved this study, and patient-informed consent was obtained of all study subjects. The diagnosis of MFS was determined according to the then current Ghent criteria15 16
Mutation classification
Sanger sequencing of the 65 coding FBN1 exons in genomic DNA from blood cells was used to detect mutations. Large deletions were tested with multiplex ligation-dependent probe amplification. All known pathogenic FBN1 mutations were classified as DN or HI as described previously predicted with the Alamut software.4 The Alamut software was shown to be precise in our previous paper confirmed byskin biopsies.4 In short, DN mutations lead to a stable mutant fibrillin-1 protein, with altered or shorter structure, which is incorporated together with the normal fibrillin-1 protein (derived from the non-mutated allele), mostly caused by missense and exon-skipping mutations.12 14 17 HI mutations lead to a reduced amount of normal non-mutated fibrillin-1 protein derived from the non-mutated allele alone, caused by whole FBN1 deletion,10 degradation of mutated messenger RNA (mRNA) by nonsense-mediated decay in frameshift and nonsense mutations11 or by protein degradation after mRNA synthesis.12 No skin biopsies were available. If certainty of the phenotypic effect was missing, they were excluded from the analysis.
Study objectives
The study aimed to determine the impact of mutation type on: (1) aortic diameters; (2) aortic dilation rate; (3) cardiovascular events (ie, aortic dissection and cardiovascular mortality).
For the first two objectives, aortic diameters were measured with the two experienced observers (RF and GT) unaware of the genotype at the level of the sinuses of Valsalva and tubular ascending aorta, aortic arch immediately prior to the left subclavian artery, proximal descending thoracic aorta and abdominal aorta. As recommended by the American Society of Cardiology (ASE)18 and European Association of Echocardiography (EAE),19 measures were made using two-dimentional imaging at end-diastole, in a strictly perpendicular plane to that of the long axis of the aorta using the leading edge to leading edge convention for aortic root, sino-tubular junction and ascending aorta; and the inner edge to inner edge convention for aortic arch, descending aorta and abdominal aorta.
Aortic dilation progression was calculated in all patients with at least 1 year of follow-up as the difference between the first and last echocardiographic follow-up study. For the third study objective, patients were followed up from the first clinic visit to the date of the first cardiovascular event, aortic dissection or cardiovascular death. Patients were censored at their last clinic visit. Aortic dissection was defined as any aortic dissection (type A or B) confirmed by an imaging modality. Cardiovascular events were updated in April 2015.
Statistical analysis
Data are presented as mean (±SD) or as number of patients (percentage). Univariate comparisons between groups were made by Student’s t-test and the χ2 test as appropriate. The aortic dilation rate was evaluated by covariance analysis with baseline aortic diameter as covariate. Interaction test between regression lines was used to analyse the significance between the inclinations of patients with HI and DN. Cox regression analysis corrected for age at baseline and gender was used to analyse the mutation effect on clinical events. All statistical tests were two sided and differences were considered statistically significant at p<0.05. Data analysis was performed using the SPSS statistical package V.20.0.
Results
Baseline characteristics
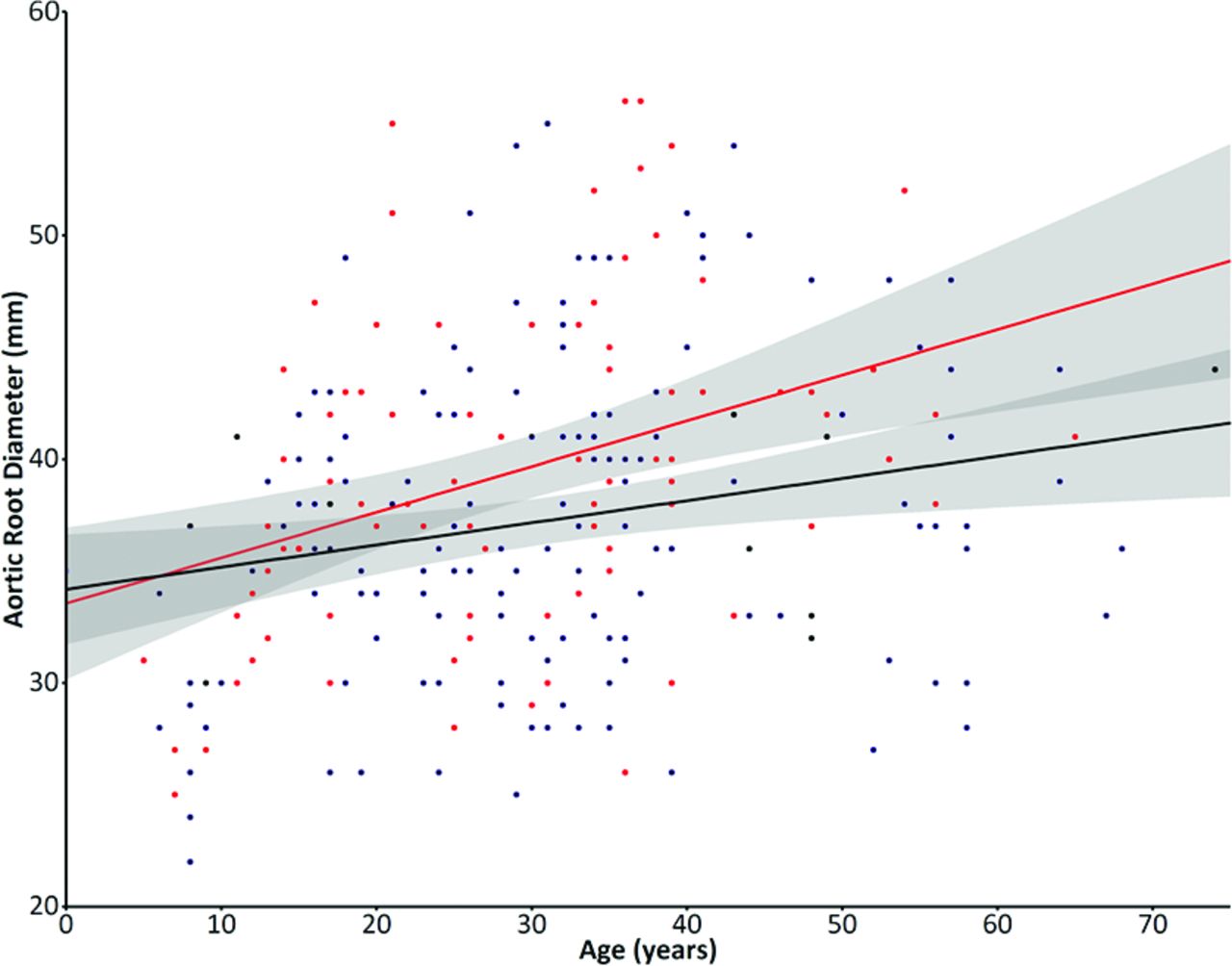
Five hundred thirty-seven patients fulfilling the Ghent criteria were followed up, 351 (65.4%) of whom underwent mutation analysis. A pathogenic FBN1 mutation was found in 312 patients (88.9%), 22 of whom could not be classified according to the classification (see online supplementary table G). Finally, 290 patients with MFS with a classified FBN1 mutation, mean age 30.2±14.7 years and sex equally distributed in the cohort with 48.5% males, were included in the analysis. A HI mutation was present in 113 (39.0%) and comprised 54 nonsense mutations, 45 frameshift mutations leading to nonsense-mediated decay, 9 missense mutations lead to unstable fibrillin-1 protein and subsequent protein degradation (n=8) or no conversion of profibrillin-1 to fibrillin-1 and thus no production of fibrillin-1 (n=1) and 5 intronic mutations leading to a splice site mutation and subsequent nonsense-mediated decay, confirmed by skin biopsies in previous studies. A DN mutation was present in 177 (61.0%) and comprised 136 missense mutations, 21 small inframe insertions or deletions, 15 inframe exon skipping mutations and 5 frameshift mutations in the nonsense-mediated decay insensitive area (see online supplementary tables A–F1). Baseline characteristics are shown in table 1. At baseline, patients with an HI mutation had a significantly larger aortic root diameter than those with a DN mutation (HI: 39.3±7.2 mm vs DN: 37.3±6.8 mm, p=0.022), with no differences in age (HI: 28.7±13.8 years vs DN: 31.1±15.2 years, p=0.191) or body surface area (HI: 1.84±0.38 m2 vs DN: 1.85±0.31 m2, p=0.841). Furthermore, at baseline, the prevalence of previous aortic surgery (HI: 18.6% vs DN: 16.3%, p=0.635) or aortic dissection (HI: 10.6% vs DN: 6.2%, p=0.186) did not differ between groups. However, age at surgery was significantly lower in patients with HI (HI: 29.9±10.1 years vs DN: 36.6±12.9 years, p=0.050) than patients with DN. No differences existed in cardiovascular treatment at baseline (HI: 53.2% vs DN: 56.1%, p=0.712). Patients with DN more frequently had ectopia lentis than patients with HI (HI: 24.8% vs DN: 52.8%, p<0.001). Patients with HI trended to have a larger aortic root diameter at the age of inclusion as compared with those with a DN mutation (B=0.105, CI –0.023 to 0.233, p=0.107, figure 1).
Baseline characteristics
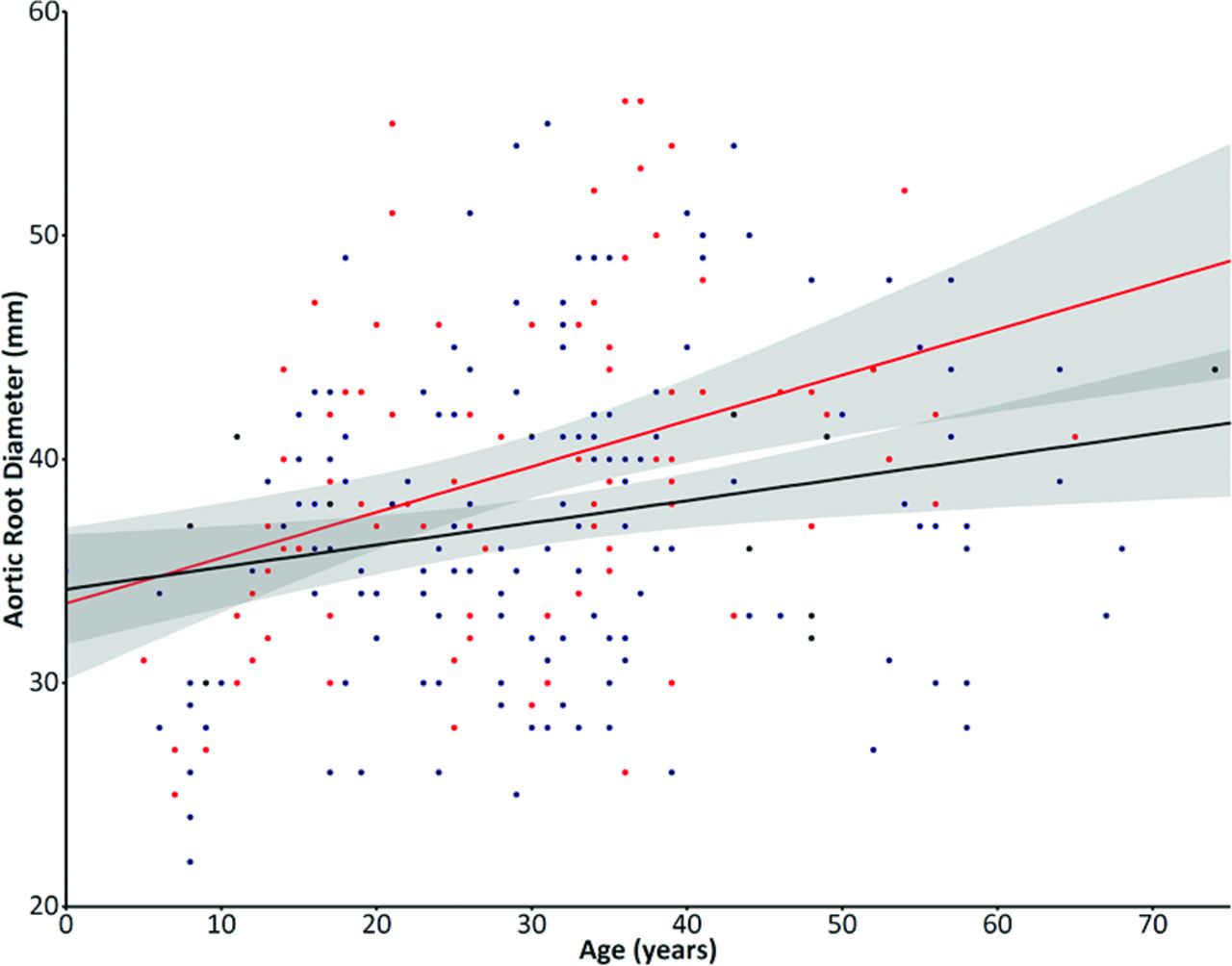
Aortic root diameters at baseline graph shows the correlation of baseline aortic root diameters with age of inclusion between patients with an haploinsufficient (HI) mutation (in red), which trended to be larger compared with those with a dominant-negative (DN) mutation (in blue). The shaded area demonstrate the CI, which is partly overlapping especially at the younger ages.
Aortic root dilation rate between genotype subgroups
The clinical follow-up data are shown in table 2. Corrected for baseline aortic diameter, the aortic dilation rate was significantly increased in patients with HI at the aortic root (HI: 0.57±0.8 vs DN: 0.28±0.5 mm/year, p=0.004) and at the tubular ascending aorta (HI: 0.59±0.9 vs DN: 0.30±0.7 mm/year, p=0.032). No differences were observed between patients with DN and HI for the arch and proximal descending thoracic and abdominal aorta (figure 2).
Aortic dilation rate between genotype subgroups

{kind=link}
{kind=link}
Aortic dilation rates box-and-whiskers demonstrate the aortic dilation rates in patients with a dominant-negative (DN) mutation (grey) and a haploinsufficient (HI) mutation (red) at the level of the sinuses of Valsalva of the aortic root (AoR), tubular ascending aorta (Asc), aortic arch (Arch), proximal descending thoracic aorta (Desc) and abdominal aorta (Abd). #p<0.01, ns=not significant.
Aortic complications between genotype subgroups
During follow-up, 5 (1.7%, mean age: 42.2±13.2 years) died, 11 (3.8%, mean age: 41.4±10.3 years) presented an aortic dissection and 55 (19.0%, mean age: 36.7±11.4 years) needed aortic surgery. Patients with HI tended to be at 3.3-fold increased risk compared with patients with a DN mutation for the combined clinical endpoint, aortic dissection and death (HI: 6.2% vs DN: 2.3%, HR: 3.3, 95% CI 1.0 to 11.4, p=0.060), corrected for age at baseline and gender. For the individual endpoints, patients with HI were not at a significantly increased risk for cardiovascular mortality (HI: 2.7% vs DN: 1.1%), aortic dissection (HI: 6.2% vs DN: 2.3%) or aortic surgery (HI: 18.6% vs DN: 17.4%).
The 290 patients with a pathogenic FBN1 mutation comprised 169 probands and their affected family members. Analysis including only the probands rendered similar results (43,3% HI with a mean aortic root dilation rate of 0.55±0.61 vs 56,7% DN with a mean aortic root dilation rate of 0.31±0.47 mm/year, p=0.026).
Discussion
This is the first study to demonstrate that patients with an HI mutation have more rapid aortic root and ascending aorta dilation rates compared with those with a DN mutation. Furthermore, HI tended to be at increased risk for dissection or cardiovascular death compared with patients with DN- FBN1 mutation as it was shown in larger Dutch national study.
Although genotype–phenotype correlations in MFS are difficult owing to the clinical variability within and between families, two genotype–phenotype correlations have been established. In a large cohort, Faivre et al showed that patients with a cysteine missense mutation (classifiable DN) are at increased risk for ectopia lentis compared with those with a premature termination codon (classifiable as HI).20 In addition, a study by Aoyama et al had already demonstrated in 1995 that mutations leading to a very low deposition of the fibrillin-1 protein (HI) were associated with shortened event-free survival and more severe cardiovascular complications.6 Moreover, among patients with Ghent-positive MFS, a higher frequency of HI-FBN1 mutations was found in patients with cardiovascular events (79%) versus patients without an event (48%, p=0.0039), even at a younger age, compared with patients with a DN-FBN1 mutation.5 Finally, we recently confirmed that patients with an HI-FBN1 mutation are at increased risk for hard clinical endpoints, including aortic dissection and death, compared with those with a DN-FBN1 mutation.15 These studies revealed intrinsic differences between MFS populations with an HI-FBN1 or DN-FBN1 mutation. The present study reproduced the results of previous studies in a different population, thereby confirming that those with an HI-FBN1 mutation had an increased dilation rate of the aortic root and ascending aorta. Furthermore, we confirmed that patients with HI-FBN1 tended to be at increased risk for the combined clinical endpoint (ie, aortic dissection and death) compared with patients with DN-FBN1. Thus, we conclude that patients with an HI-FBN1 mutation require more intense surveillance with a stricter imaging protocol to at least yearly image of the aortic root and a low threshold to perform an extra echocardiography. It is unknown why patients with an HI-FBN1 mutation are at increased cardiovascular risk compared with patients DN-FBN1. Interestingly, Aubart et al demonstrated a fourfold variation in FBN1 mRNA synthesis levels between controls without MFS with a very stable 50/50 allelic contribution,7 and a similar FBN1-mRNA variation was found in patients with MFS with an HI-FBN1 mutation; however, the level of FBN1 mRNA was half that of the controls and >90% was transcribed from the non-mutated allele, thereby confirming the concept of HI. Finally, the authors showed a low level of FBN1 mRNA to be an important determinant for phenotype severity and tends to raise the risk of aortic dilation.7 The present study reveals interesting but not very discriminatory results. A low amount of fibrillin-1 protein, due to epigenetics or environmental influences, seems to lead to the more severe phenotype.
The reason for concerted mRNA expression of the mutated and non-mutated allele in MFS remains unknown. However, this may give rise to interesting novel therapeutic options for patients with HI-FBN1, for example, stimulation of the non-mutated allele by FBN1 expression regulators located outside the FBN1 locus.7 In addition, in a substudy of the COMPARE (COzaar in Marfan PAtients Reduces aortic Enlargement) study, patients with HI-FBN1 responded to the addition of losartan to beta-blockers, with a significant reduction in the aortic dilation rate.4
The present retrospective study has several limitations. First, mutation analysis was performed in only 64% of patients in this study; others refused mutation analysis or analysis was not considered necessary for diagnosis and counselling. Second, our classification was based on software programming, knowledge of previous mutations and the literature. Skin biopsies to test protein stability and the effect on mRNA were not available. Third, few patients died or had an aortic dissection during follow-up; thus, the significance on the combined clinical endpoint was based on a small number of patients. In addition, the small number of events translates to the lack of power to show significance between the type of mutation and the individual endpoints.
In conclusion, patients with an HI mutation had a more severely affected aortic phenotype, with larger aortic root diameters, a more rapid dilation rate and tended to have an increased risk of death and dissections compared with patients with a DN mutation.
Key messages
What is already known on this subject?
MFS is an autosomal-dominant connective tissue disorder caused by FBN1 mutations with multisystemic involvement. Phenotypic expression and age at manifestations onset are known to vary widely in patients with MFS. Previous studies have reported different effects of FBN1 mutations classified as dominant negative (DN) or haploinsufficiency (HI) on the severity of cardiovascular manifestations.
What might this study add?
This is the first longitudinal study with echocardiographic follow-up data in patients with MFS classified into HI versus DN, demonstrating that patients with HI had a larger aortic root diameter at baseline and a faster aortic root dilation rate during follow-up (HI: 0.57±0.8 vs DN: 0.28±0.5 mm/year, p=0.004) compared with patients with DN.
Furthermore, this is the second longitudinal study confirming that patients with HI tended to have an increased risk for dissection and death compared with patients with DN.
How might this impact on clinical practice?
Aortic dilation rate and aortic events in patients with MFS are at least partially dependent on mutation type. We conclude that patients with an HI-FBN1 mutation require more intense surveillance with a stricter imaging protocol. Larger multicentre studies are required to confirm these findings and to optimise treatment strategies between these groups.
References
Footnotes
Funding This study has been funded by Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo (CIBERCV), Spain through the project PI14/01062 (Co-funded by European Regional Development Fund). The author RF is funded by a fellowship grant from the Interuniversity Cardiology Institute of the Netherlands. Authors declare no relationships with industry.
Competing interests None declared.
Patient consent Obtained.
Ethics approval METC Barcelona.
Provenance and peer review Not commissioned; externally peer reviewed.