Article Text

Statistics from Altmetric.com
Despite significant strides towards an understanding of the initiation and progression of atherosclerosis and the influence of risk factors, coronary heart disease remains the principal killer in the western world.1 If progress in the field is to continue in the 21st century, one must focus on high risk patients with lesions that are vulnerable to thrombosis together with the triggering mechanisms that cause plaques to rupture at a precise location and time. Although animal studies have helped define the molecular mechanisms of atherosclerosis, a convincing model of plaque rupture still does not exist. Therefore, the development of future treatments targeted against plaque instability is contingent upon our ability to confidently recognise precursor lesions likely to thrombose; this will be primarily achieved via improved imaging modalities.
Insights into the mechanisms of coronary thrombosis extend from detailed analyses of underlying plaque morphologies in necropsy specimens from sudden death victims.2,3 In 50–75% of these cases, the culprit lesion (fatal plaque) shows a coronary thrombus whereas the remainder without thrombi exhibit stable coronary plaques with > 75% cross sectional area luminal narrowing.4 The major cause of acute coronary thrombosis is plaque rupture, and the precursor lesion has been termed vulnerable plaque (fig 1) or, as defined by our laboratory, the thin cap fibroatheroma (TCFA). In this review, we will critically analyse the pathology of plaque rupture with emphasis on its relation to TCFAs and healed plaque ruptures to gain a better understanding of the lesion most responsible for coronary morbidity and mortality.

A non-haemodynamically limiting thin cap fibroatheroma. (A) Low power view of a thin cap fibroatheroma (TCFA) with a relatively large eccentric necrotic core (NC) and an overlying thin fibrous cap (< 65 μm); the greyish material in the lumen is barium gelatin (Movat pentachrome, ×20). (B) Image of the boxed area in panel A showing an advanced necrotic core with a loss of matrix containing numerous cholesterol clefts and cellular debris (×100). (C) High power view of a fibrous cap heavily infiltrated by inflammatory cells (haematoxylin and eosin, ×200). (D) Numerous CD-68 positive macrophages are localised to the fibrous cap (×200). (E) The fibrous cap shows rare α-actin positive smooth muscle cells (SMCs) (×200).
ACUTE CORONARY SYNDROMES AND THROMBOSIS
Patients with acute coronary syndromes classically present with unstable angina, acute myocardial infarction, or sudden coronary death. Most acute coronary syndromes are precipitated by luminal thrombi, which arise from three different plaque morphologies: rupture, erosion, and calcified nodules.4 Of these, plaque rupture is the most frequent, accounting for 60–75% of cases. Plaque rupture develops in a lesion with a necrotic core and an overlying thin disrupted fibrous cap heavily infiltrated by macrophages and T lymphocytes; a luminal thrombus develops because of physical contact between platelets and the thrombogenic necrotic core. In contrast, erosions are characterised by a luminal thrombus superimposed on a proteoglycan-rich plaque containing mostly smooth muscle cells with few inflammatory cells; these plaques account for 25–40% of all coronary thrombi.4–6 Most eroded lesions lack a necrotic core, but when present, there is no direct communication with the luminal thrombus since the overlying fibrous cap is typically thick and intact. Finally, the calcified nodule is the least common (2–7%) of all lesions that cause coronary thrombi.4,5 These lesions typically contain calcified plates along with bony nodules that penetrate the lumen, which contains disrupted endothelium.
PLAQUE RUPTURE AND ITS PRECURSOR LESION:THE THIN CAP FIBROATHEROMA
Ruptured plaques typically contain a relatively large necrotic core with an overlying thin disrupted fibrous cap infiltrated by macrophages and T cells. The smooth muscle cell content within the cap is generally absent or sparse. The thickness of the fibrous cap near the rupture site approximates 23±19 μm, with 95% of the caps measuring less than 65 μm.2 Those plaques that closely resemble ruptures (necrotic cores with thin caps) but lack a luminal thrombus have been designated by our laboratory as TCFA or, more traditionally, vulnerable plaques (fig 1 and 2).4 In a more conservative sense, however, the term “vulnerable” should be used for lesions that underlie all causes of coronary thrombi, including pathologic intimal thickening, thick and thin cap fibroatheromas, and calcified plaques with nodules.
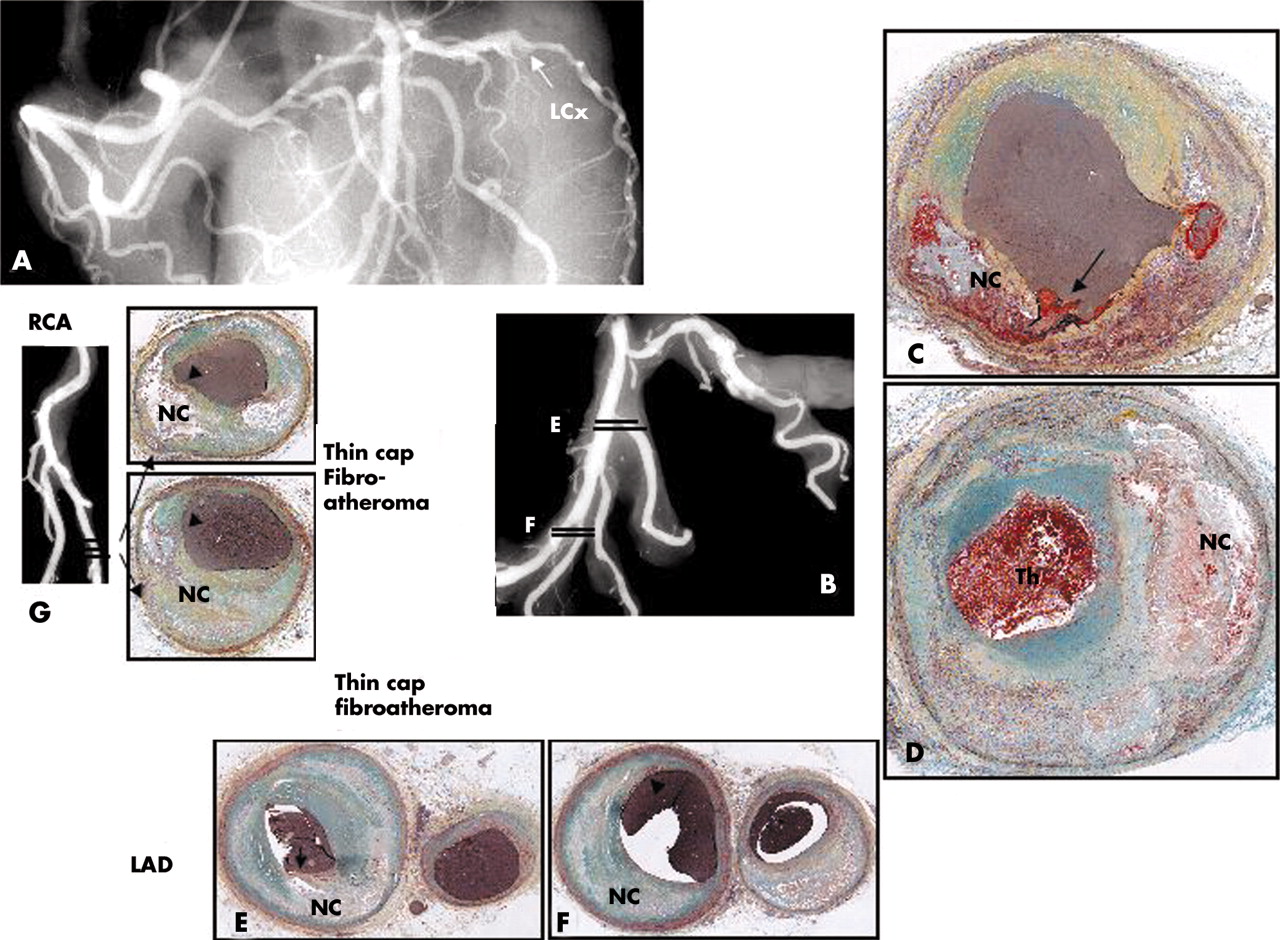
Plaque rupture in a 43 year old white male who collapsed at work and could not be resuscitated. The patient had recent complaints of shoulder pain and headache but no known medical history or risk factors. At necropsy there was haemopericardium with 500 ml of blood and a long vertical tear on the posterolateral surface of the left ventricle. There was an acute transmural myocardial infarction in the lateral wall of the left ventricle and a haemorrhagic tract in the area of the rupture, which was located in the middle of the infarct. The myocardium showed coagulation necrosis with prominent neutrophilic infiltrate consistent with a 2–3 day old infarct. A postmortem angiogram showed total occlusion of the left circumflex (LCx) artery (arrow in A and B). Histologic sections demonstrate fibrous cap rupture (arrow in C) with underlying hemorrhagic necrosis (NC). (D) Just distal to the site of rupture the coronary artery there is approximately 70% diameter stenosis and an overlying occlusive thrombus. (E) Sections of the first diagonal and LAD (E) and the distal third diagonal (F) show fibrous cap thinning with mild insignificant luminal narrowing and positive remodelling. (G) The angiogram of the distal right coronary artery (RCA) and the PDA shows mild irregularities at the site of sectioning, shown in dark lines. Note sites of fibrous cap thinning (arrows) overlying relatively larger necrotic cores (NC).
Although there are similarities, TCFAs differ from ruptured plaques based on a trend towards a smaller necrotic core, fewer macrophages within the fibrous cap, and less calcification (table1). Several morphologic features in various human coronary plaque types, including culprit lesions, are shown in table 2. Overall, cross sectional luminal narrowing is greater in ruptures than in TCFAs, with occlusive thrombi having greater underlying stenosis than non-occlusive thrombi (figs 3 and 4). The number of cholesterol clefts in the necrotic core, vasa vasorum, and macrophages are significantly greater in ruptured than eroded plaques or stenotic stable lesions. Macrophage content is also lower in TCFAs compared with ruptures, which also have a greater accumulation of hemosiderin.
Morphologic characteristics of plaque rupture and thin-cap fibroatheroma
Comparison of necrotic core size, number of cholesterol clefts, macrophage infiltration, number of vasa vasorum, and haemosiderin laden macrophages in culprit plaques

Morphometric analysis of thin-cap fibroatheromas. (A) Shows the percentage of cross sectional area luminal narrowing by plaque type. The thin cap fibroatheromas (TCFAs) and fibroatheromas are less narrowed than acute or healed ruptures. (B) The percentage of TCFAs are plotted against the degree of luminal narrowing. Notably, over 80% of these lesions have < 75% cross sectional area luminal narrowing.

Healed plaque rupture. (A) Areas of intraintimal lipid-rich core with haemorrhage and cholesterol clefts; an old area of necrosis (NC) is seen underlying a healed thrombus (HTh). (B) Higher magnification showing extensive smooth muscle cells (SMCs) within a collagenous proteoglycan-rich neointima (healed thrombus) with clear demarcation from the fibrous region of old plaque to right. (C and D) Layers of collagen by Sirius red staining. (C) Note area of dense, dark red collagen surrounding lipid haemorrhagic cores seen in corresponding view in A. (D) Image taken with polarised light. Dense collagen (type 1) that forms fibrous cap is lighter reddish yellow and is disrupted (arrow), with newer greenish type III collagen on right and above rupture site. (A and B) Movat pentachrome. Reproduced with permission from Burke et al, Circulation 2001;103:934–40.
Intraplaque haemorrhage in other sites of the coronary vasculature is more common in hearts with ruptures versus stable plaques without luminal thrombi. The mean (SD) number of acute haemorrhages in non-culprit lesions from patients with rupture was 2.5 (1.3) versus none in erosion (p = 0.0001) and 0.05 (0.6) in stable plaques (p = 0.04).4 Evidence of prior haemorrhages as analysed by anti-glycophorin A staining (a protein specific to erythrocyte membranes) was significantly greater in TCFAs than in fibroatheromas with early or late necrotic cores or lesions with pathologic intimal thickening. The degree of glycophorin A staining correlated with both necrotic core size and the extent of macrophage infiltration suggesting that erythocytes may directly contribute to necrotic core expansion and lesion instability.7
HEALED PLAQUE RUPTURE AND PROGRESSION OF ARTERIAL STENOSIS
Morphologic studies suggest that plaque progression beyond 40–50% cross sectional luminal narrowing occurs secondary to repeated ruptures, which may be clinically silent.8,9 Healed plaque ruptures are detected microscopically by the identification of breaks in the fibrous cap (type I collagen) with an overlying repair reaction consisting of a proteoglycan-rich mass, collagen type III, and interspersed smooth muscle cells (fig 5).8 Late healed ruptures show less accumulated proteoglycans and the recurrence of type I collagen.

Luminal narrowing as a function of plaque morphology. (A) Severe cross sectional luminal narrowing of > 75% is more common in acute and healed ruptures than in thin cap fibroatheromas (TCFAs). Note 74% of TCFAs showed luminal narrowing ⩽ 74%. (B) Cross sectional luminal narrowing in lesions with occlusive and non-occlusive thrombi; note lesions with occlusive thrombi demonstrate greater luminal stenosis. (C) The change in lumen size is plotted against the number of healed ruptures. While the percentage of stenosis gradually increases with 1–3 underlying healed ruptures, with four previous ruptures, there is a pronounced increase in luminal stenosis. Panel A is reproduced with permission from Virmani R, et al. J Interv Cardiol 2002;15:439–46.
The underlying mechanisms of atherosclerotic stenosis are poorly understood. The prevalence of silent ruptures in the general population is, by definition, unknown and few angiographic studies have demonstrated plaque progression. As Davies initially observed, the frequency of healed plaque ruptures parallels an increase in luminal narrowing.9 In plaques with 0–20% diameter stenosis, the incidence of healed plaque ruptures was 16%, for lesions with 21–50% stenosis, 19%, and plaques with > 50% narrowing, 73%. In our experience, 61% of hearts from sudden coronary death victims have healed plaque ruptures and, comparable to the study by Davis, the percentage of stenosis increases with the number of healed plaque ruptures. Multiple healed ruptures with proteoglycan and collagen layering are common in segments with acute and healed ruptures. The underlying percentage luminal narrowing for acute ruptures exceeds that for healed ruptures (79 (15)% v 66 (14)%, p = 0.0001),8 suggesting that stabilisation of vulnerable plaques may prevent the likelihood of repeated ruptures, thereby greatly reducing or abolishing the potential for future ischaemic events.
MORPHOLOGIC CHARACTERISTICS OF PLAQUES THAT RUPTURE
The extent of luminal narrowing varies with underlying plaque morphology. TCFAs and fibroatheromas have the least luminal narrowing while lesions with acute plaque rupture, haemorrhage, or healed repair sites show the most stenosis. Over 74% of TCFAs have < 75% cross sectional area luminal narrowing (equivalent to < 50% diameter stenosis). Healed and acute plaque ruptures show the greatest luminal narrowing with 46% and 43%, respectively, containing > 75% cross sectional area narrowing. In contrast, only 26% of TCFAs show severe luminal narrowing (fig 3B). In a population where sudden cardiac death is the first manifestation of coronary disease, these morphologic findings strongly suggest that TCFAs are precursors to acute ruptures and healed plaque ruptures are often clinically silent.
TCFAs with ⩽ 50% diameter stenosis may be the ideal candidate lesions one should first attempt to identify and treat as a measure to reduce the incidence of myocardial infarction in high risk patients. If a drug eluting stent becomes the treatment choice for lesions with insignificant plaque burden (< 50% diameter stenosis), the goal should be an increase in diameter < 20% since with each silent healed rupture, the increase in stenosis is only approximately 20% (fig 4C). Moreover, the location and distribution of multiple TCFAs may also influence the decision not to treat or not to treat with a drug eluting stent. Currently, systemic pharmacologic treatment is likely to be more desirable for the stabilisation of TCFAs; as shown in animal studies, these agents have the potential to reduce necrotic core size as well as macrophage infiltration.10–12
The mean necrotic core size in lesions from sudden coronary death victims is independent of luminal narrowing and is greatest in plaque ruptures, followed by TCFAs, and fibroatheromas (table 3). Conversely, the length of the necrotic core in ruptures and TCFAs is similar, varying from 2–22.5 mm, with a mean of 8 and 9 mm, respectively (table 3).13 In determining the instability of a plaque, it may be critical whether the necrotic core is circumferential. In TCFAs, 75% of lesions show a necrotic core circumference of > 120° (RV, unpublished data). Serial sectioning through these lesions shows an extensive heterogeneity in plaque morphology in relation to the position of the necrotic core within the lesion. As illustrated in fig 6, within a short millimetre distance the atherosclerotic plaque may vary in its presentation with a necrotic core infringing on the lumen forming a TCFA, while a short distance away, the same necrotic core now lies deep from a superficial healed repair site. The acute plaque rupture may be present alone or away from the core. Therefore it is possible to find healed rupture sites with multiple acute ruptures and vulnerable TCFAs in a relatively short coronary segment. This degree of plaque complexity may render treatment with a drug eluting stent ineffective or even dangerous because delayed healing may further contribute to plaque instability.
Approximate size of the necrotic core in advanced plaques

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serial sections of the left circumflex coronary artery from the case shown in fig 2D demonstrating sites of acute and healed plaque rupture and thin cap fibroatheromas (TCFAs) within a relatively short distance. The numbers in the lower right hand corner of the micrographs correspond to the distance in mm from the previous section within a single 5 mm arterial segment. The arrows correspond to rupture sites whether healed or acute while arrowheads represent sites of TCFAs. The first section (A) at 0.2 mm shows a healed plaque rupture; the circumferential repair is rich is proteoglycans (bluish-green). Overall the vessel shows multiple sites of rupture at depths of 2.7 mm and 3.0 mm; an occlusive luminal thrombus is observed in most sections except for panels J–L where the lumen is filled with barium gelatin (greyish material). In the same artery at levels of 3.3 mm, 3.6 mm, and 3.9 mm are multiple TCFAs. Note similar areas of necrosis are found in both deep and superficial locations depending on the level of the section. (Movat pentachrome, ×20).
INCIDENCE OF THIN CAP FIBROATHEROMA IN VARIOUS CORONARY SYNDROMES, DISTRIBUTION AND EXTENT OF MACROPHAGE INFILTRATION
In patients with an acute myocardial infarction, the incidence of TCFAs is highest in males with a mean of 3 per heart, with half as many in women. In patients dying suddenly, however, the incidence is similar between both sexes. Incidental deaths or those from plaque erosion show the fewest number of TCFAs. The number of TCFAs are also lower in patients dying with healed plaque ruptures or stable plaques.8
The majority of TCFAs and acute and healed ruptures occur in the proximal portion of the three major coronary arteries; less than half arise in the mid portion and few are found in the distal vessels. The proximal left anterior descending coronary artery is the most frequent location with proximal right and left circumflex coronary arteries about half as common.
Our laboratory has shown that fibrous cap thickness is dependent on the extent of macrophage infiltrate; the thicker the fibrous cap the fewer the macrophages. In serial sections of TCFAs, the necrotic core may be localised deep within the plaque, but over a relatively short distance of < 1.4 cm, the lipid core may approach the lumen. Therefore, histologically the presence of suspected TCFAs may be better identified through serial sectioning.
ROLE OF RISK FACTORS IN PREDICTING THIN CAP ATHEROMAS
TCFAs are a frequent finding in men dying suddenly with coronary thrombosis, in particular in individuals with a high total cholesterol (TC) and TC/high density lipoprotein (HDL) ratio (> 210 mg/dl (>5.38 mmol/l) and TC/HDL-C ratio > 5, respectively).2 The incidence of TCFAs in women is most frequent in those over 50 years of age with total cholesterol > 210 mg/dl (>5.38 mmol/l).3 Another risk factor reported to predict the development of acute coronary syndromes is high sensitivity C reactive protein (hs-CRP).14 The increased relative risk of sudden cardiac death associated with hs-CRP is seen only in the highest quartile, in which individuals are at a 2.78-fold increase in risk of sudden cardiac death (95% confidence interval 1.35 to 5.72) compared to men in the lowest quartile. In addition, the mean number of TCFAs was most frequent in patients with high serum hs-CRP than in those with lower hs-CRP values.14
LESSONS FROM PATHOLOGY FOR THE INTERVENTIONALIST
Several recent clinical angiographic and intravascular ultrasound (IVUS) studies report multiple complex coronary plaques in patients with acute myocardial infarction with ST segment elevation. The concept of multifocal plaque instability is further supported by angiographic natural history studies in patients with acute myocardial infarction, in whom rapid progression of both culprit and non-culprit lesions over a period of one month has been documented. The significance of this finding is vital to the patient’s clinical outcome since two fifths of the 253 individuals diagnosed with multiple complex coronary plaques reported by Goldstein et al had a less than favourable hospital course. The pathologic results in necropsy specimens of culprit and non-culprit plaques are in close agreement with the clinical observations. Independent of advanced imaging techniques, the interventionalist, with reasonable certainty, should be able to identify high risk non-culprit plaques based on the coronary pathology of sudden death patients. For example, patients with cholesterol ratios > 5 with hs-CRP in the fourth quartile and elevated values of other surrogate biomarkers of sudden death such as MMP-9 or MCP-1 should be considered at high risk for unstable plaques (table 4). This, together with the finding of proximal coronary lesions of > 50% diameter stenosis with necrotic cores > 120° would strongly suggest the presence of plaque with a vulnerable morphology.
Clinical variables for the identification of non-culprit lesions at risk for rupture in high risk patients
SUMMARY AND CONCLUSIONS
The majority of acute coronary syndromes are the result of plaque rupture. The lesion that mostly resembles acute rupture is the TCFA, which is characterised by a necrotic core with an overlying fibrous cap measuring < 65 μm. These lesions contain rare smooth muscle cells and numerous macrophages; the presence of T lymphocytes is variable. TCFAs are most frequent in patients dying with acute myocardial infarction and are least common in incidental non-coronary deaths. They are primarily located in the proximal coronary arteries, in particular the left anterior descending. The average necrotic core length is 2–17 mm (mean 8 mm) and the average cross sectional area narrowing in the majority of lesions is < 75%. Only 26% of TCFA have > 50% diameter stenosis, whereas approximately 50% of healed plaque ruptures and acute ruptures show severe stenosis. It is important to emphasise that complex lesions such as TCFAs show a great degree of heterogeneity over a relatively short distance. Serial sectioning may reveal multiple healed plaque ruptures, TFCAs, or acute rupture sites.
Coronary risk factors for TCFAs are high TC and high TC/HDL-C ratio, women > 50 years, and patients with elevated values of hs-CRP. Further, patients dying with acute myocardial infarction are likely to have multiple TCFAs versus those dying from severe coronary stenosis in the absence of acute thrombi or incidental death. Because of its clinical significance, the identification of TCFAs will play a pivotal role towards reducing the morbidity and mortality of coronary artery disease. The complex nature of TCFAs dictates the placement of a drug eluting stent only in lesions with ⩾ 50% diameter stenosis while systemic therapy may be a more logical approach for treating insignificantly narrowed TCFAs.
Acknowledgments
Support in part by a research grant from the National Institutes of Health (RO1 HL61799–02). We are indebted to Hedwig Avallone and Lila Adams (Armed Forces Institute of Pathology) for their excellent technical assistance.
REFERENCES
Footnotes
-
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or reflecting the views of the Department of the Army, the Department of the Air Force, or the Department of Defense.
Linked Articles
- Miscellanea
- Miscellanea