Article Text

Abstract
Background Tetralogy of Fallot (TOF) is common in individuals with hemizygous deletions of chromosome 22q11.2 that remove the cardiac transcription factor TBX1.
Objective To assess the contribution of common and rare TBX1 genetic variants to TOF.
Design Rare TBX1 variants were sought by resequencing coding exons and splice-site boundaries. Common TBX1 variants were investigated by genotyping 20 haplotype-tagging SNPs capturing all the common variations present at the locus. Association analysis was performed using the program UNPHASED.
Patients TBX1 exons were sequenced in 93 patients with non-syndromic TOF. Single nucleotide polymorphism analysis was performed in 356 patients with TOF, their parents and healthy controls.
Results Three novel variants not present in 1000 chromosomes from healthy ethnically matched controls were identified. One of these variants, an in-frame 57 base-pair deletion in the third exon which removed 19 evolutionarily conserved residues, decreased transcriptional activity by 40% in a dual luciferase assay (p=0.008). Protein expression studies demonstrated that this mutation affected TBX1 protein stability. After correction for multiple comparisons, no significant associations between common genetic variants and TOF susceptibility were found.
Conclusion This study demonstrates that rare TBX1 variants with functional consequences are present in a small proportion of non-syndromic TOF.
- TBX1
- heart defects, congenital
- tetralogy of Fallot
- genetic association studies
- mutation
- genetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
Congenital heart defects (CHD) occur in approximately seven out of every 1000 live births.1 Tetralogy of Fallot (TOF), which accounts for approximately 7% of CHD and is the commonest cyanotic CHD,2 occurs in a number of syndromes but in the majority of cases is an isolated defect. Chromosome 22q11.2 deletions account for approximately 7% of TOF cases3 4 and trisomy 21 accounts for a further 5% of cases.3 A rarer syndromic association is Alagille syndrome which results from JAG1 haploinsufficiency or intragenic mutation. The advent of array-based molecular karyotyping has shown that approximately another 10% of cases are the result of copy number variants.5 However, around 75% of cases are unexplained and are thought to result from a complex interaction of environmental and genetic factors. In support of this model, substantial genetic influences have been inferred for certain malformations in studies of familial recurrence risk ascertained through non-syndromic patients.6 A CHD risk of 3.1% in offspring and 2.2% in siblings of patients with TOF has been reported, indicating a substantial increase in CHD risk for first-degree relatives of cases compared with the general population.6
CHD is common in patients with chromosome 22q11.2 deletion, with 17% of patients with 22q11.2 deletions having TOF.7 The deletion, which is mediated by non-homologous recombination between low copy repeats, typically removes a genomic segment including the cardiac transcription factor TBX1. TBX1-expressing progenitors contribute to the majority of the outflow tract (OFT) and right ventricle of the developing heart.8–13 Studies in mouse models have shown that Tbx1 deletions or null mutations in the mouse cause CHD including OFT lesions.8 10 14 15 Human studies have identified nine novel variants of TBX1 that alter protein sequence in patients who have clinical features of the 22q11.2 deletion syndrome, including CHD, but who do not carry a chromosomal microdeletion.16–19 Some of these mutations completely ablate TBX1 function in vitro, while others result in a gain of TBX1 function, suggesting an optimal range of TBX1 activity above or below which the risk of malformations increases.
We hypothesised that hypomorphic alleles of TBX1 which reduce but do not completely ablate TBX1 function, might be involved in susceptibility to non-syndromic TOF. Variants affecting TBX1 expression levels and thus potentially predisposing to TOF risk, could be rare or common; no previous study has investigated the role of common single nucleotide polymorphisms (SNPs) in the TBX1 gene in TOF susceptibility. In this study, we screened patients with non-syndromic TOF for rare genetic variants in all coding exons of TBX1 by resequencing and performed association analysis of common haplotype-tagging SNPs (htSNPs) in and around TBX1 in trio families, cases and controls.
Material and methods
Study population
Patients with TOF, of White European ancestry, were recruited from four UK paediatric cardiology centres. Clinical records were reviewed before recruitment, and probands with known chromosomal abnormalities, other recognised syndromes, learning difficulties, or known maternal exposure to significant teratogens during pregnancy were excluded. Parental samples were obtained where possible. Proband samples were screened for 22q11.2 deletion by multiplex ligation-dependent probe amplification (MRC-Holland, Amsterdam, Netherlands), and samples with deletions were excluded from further analysis. Ethical approval was given for the study and fully informed consent was obtained from all participants (or their parents, if children were too young to themselves consent). DNA was extracted from blood or saliva samples using standard protocols.
Exon sequencing
TBX1 coding sequence and consensus splice sites were sequenced in 93 unrelated TOF probands. Intronic PCR primers were designed based on transcripts NM_080646, NM_005992 and NM_080647 (www.ncbi.nlm.nih.gov, accessed 26 July 2010). PCR products were cleaned before bi-directional dideoxy sequencing and sequence traces analysed using the Staden Package suite of programs (http://staden.sourceforge.net/, accessed 26 July 2010). Previously unreported variants found in the TOF probands were genotyped in 1000 control chromosomes.
Constructs and luciferase assays
The TBX1 variants c.115G→A and c.129_185del57 were introduced into the TBX1 expression construct TBX1-pcDNA318 by site-directed mutagenesis. TBX1 constructs were transfected into U2-OS cells along with pGL2tk-2xT, which contains two copies of the T-binding site adjacent to firefly luciferase,19 and Renilla luciferase control. RNA was extracted from transfected cells using standard techniques, DNAase treated to avoid amplification of remaining transfected plasmid and Taqman relative-quantification real-time PCR (Applied Biosystems, California, USA) performed to check that equivalent levels of transcript were obtained for TBX1 and the TBX1 variants. The transcriptional activity of TBX1 and the TBX1 variants was then measured by comparison of the firefly luciferase reading with the Renilla luciferase control (dual luciferase assay).
Immunocytochemistry
HEK293 cells were seeded on glass cover slips and transfected with wild-type or variant TBX1. Twenty-four hours after transfection, cells were fixed, permeabilised with 0.1% Triton X-100 in phosphate-buffered saline (PBS) and after blocking with 5% goat serum in PBST probed with rabbit anti-Tbx1 antibody (Zymed) followed by the secondary antibody (Cy3 conjugated sheep anti-rabbit antibody, Sigma). Cells were mounted with VectaShield containing 4,6-diamino-2-phenylindole.
Protein expression and stability
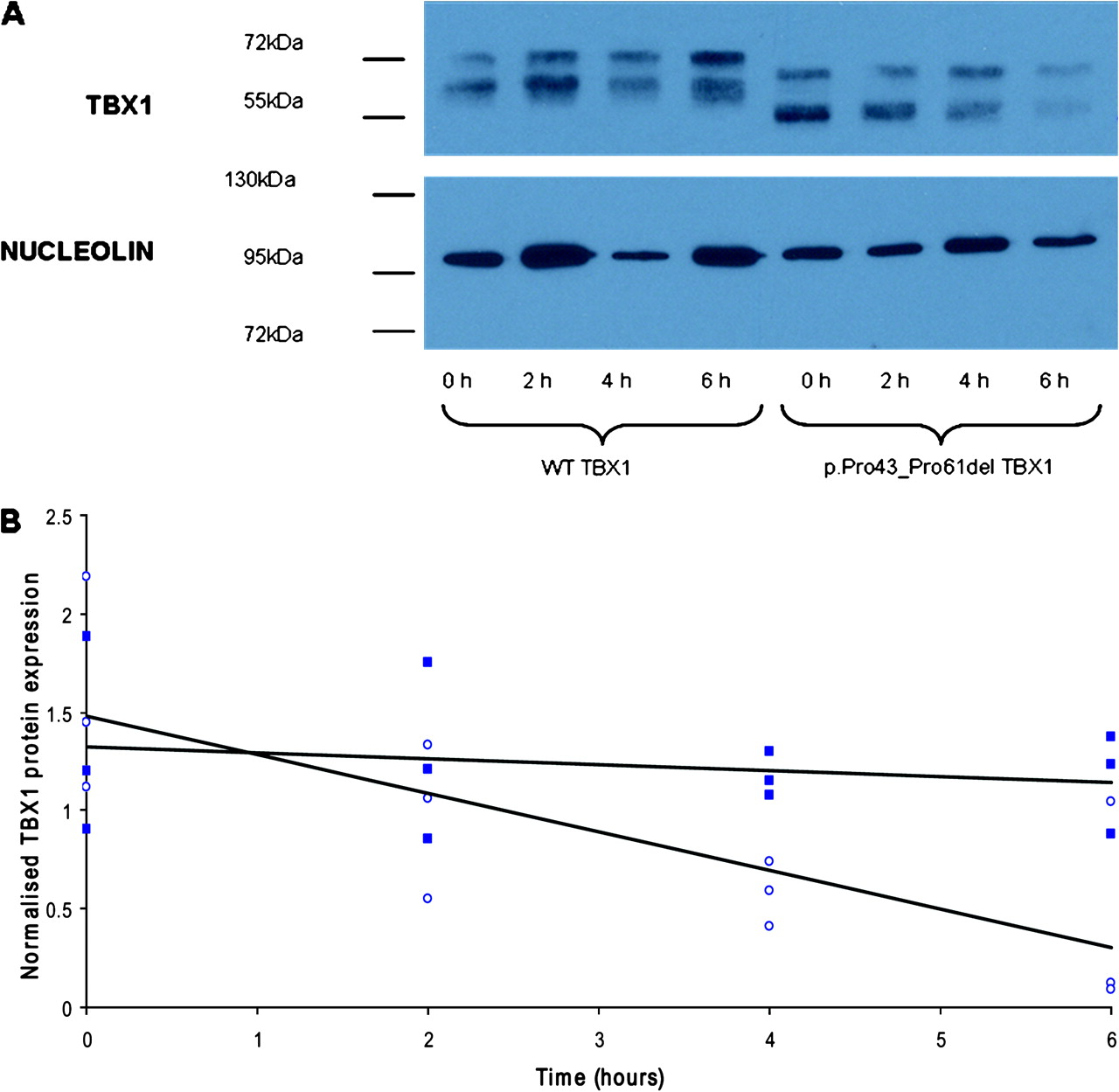
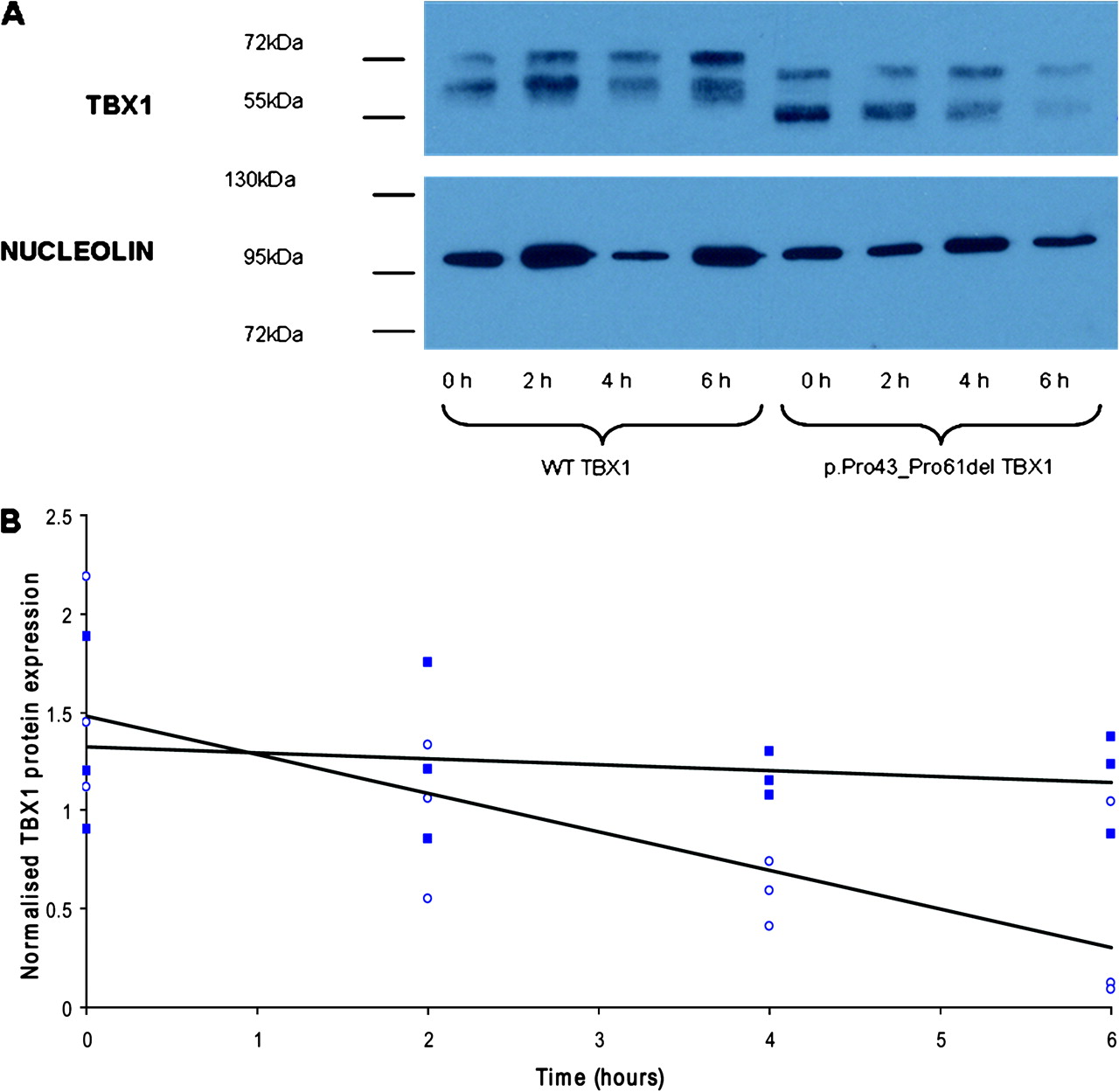
The stability of the p.Pro43_Pro61del variant protein was compared with the wild-type protein using cycloheximide inhibition of de novo protein synthesis followed by western blot analysis. HEK293 cells were transfected with wild-type or variant TBX1. Twenty-four hours after transfection, cells were treated with 25 μg/ml cycloheximide and lysed at 2 h intervals using 2X sodium dodecyl sulphate denaturing buffer (0.5 M Tris pH 6.8, 4% sodium dodecyl sulphate (w/v), 20% glycerol, 1.5% dithiothreitol (w/v)). Proteins were resolved on a 12% polyacrylamide gel and transferred onto Hybond ECL membrane (GE, Buckinghamshire, UK). Membranes were probed with anti-Tbx1 antibody (Zymed) and anti-nucleolin antibody (Bethyl, Montgomery, TX, USA) as a loading control then detected with horseradish peroxidase-conjugated anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA, USA) using West Dura ECL kit (Thermoscientific, Rockford, IL, USA). Band intensities were measured in Adobe Photoshop, normalising TBX1 expression to the loading control.
SNP genotyping
Three hundred and fifty-six patients with TOF, comprising 203 parent–child trios, 80 parent–child duos, 67 singleton probands and six probands from three multiplex families were genotyped. One hundred and eighty-two unrelated healthy individuals, free of CHD, of White European ancestry were also genotyped as controls for those cases where family members were not available. Sixteen htSNPs were selected from 15 kb upstream of TBX1 to 4.5 kb downstream of TBX1 using the HapMap data for the samples of Northern and Western European ancestry (CEU samples; http://www.hapmap.org, accessed 27 July 2010) and the Tagger utility of Haploview v3.2 (http://www.broad.mit.edu/mpg/haploview, accessed 27 July 2010). These htSNPs had a minor allele frequency >0.05 and were in linkage disequilibrium at r2 >0.8 with all other genetic variations within the region. Four additional SNPs (rs5748418, rs737868, rs13054377 and c.1189A→C), which were identified by sequencing of the TOF cases and which were not in linkage disequilibrium (R2<0.8) with the selected ht SNPs, were also included.
Statistical analysis
Luciferase activation by control and mutant TBX1 proteins and mRNA production from the different TBX1 constructs in cell culture, were compared using paired t tests. TBX1 wild-type and the p.Pro43_Pro61del variant protein concentration after cycloheximide treatment was analysed using analysis of variance (assuming a general linear model) and regression analysis in Minitab 15. Genotype data were checked for Mendelian consistency within the families and for Hardy–Weinberg equilibrium using PEDSTATS.20 Association analysis of the SNP genotypes within the families and unrelated controls was performed using UNPHASED version 3.0.9.21 UNPHASED was chosen in view of the program's capacity to handle datasets including both trio families and unrelated cases and controls and carry out genotype and haplotype association tests yielding p values for the entire dataset.
Results
Sequencing TBX1 in probands
Sequencing 93 TOF probands identified four previously unreported genetic variants present in eight probands. Three novel variants (c.115G→A, c.129_185del57 and c.1074G→A), were each present in one proband and absent from 1000 control chromosomes. The single-base substitution c.115G→A, resulting in a p.Gly39Ser amino acid substitution in exon 3 of TBX1, was inherited from a phenotypically normal mother. The patient with the 57 bp deletion (c.129_185del57) resulting in the in-frame amino acid deletion p.Pro43_Pro61del was noted to have a right-sided aortic arch. He inherited the change from his mother who has no history of congenital heart disease. A synonymous base substitution c.1074G→A was present in the exon 9C major isoform (NM_080647 isoform C) of a third proband and was also inherited from a phenotypically normal mother. The fourth previously unreported variant was a synonymous base substitution c.75G→T, p.Gly25Gly, present in five probands and seen in 20 of 1000 control chromosomes. These synonymous variants were thought unlikely to be causative and were not investigated further.
The glycine of the assumed amino acid substitution p.Gly39Ser is conserved in chimpanzee, rat, mouse and opossum (Monodelphis domestica) but is substituted by serine in the frog (Xenopus tropicalis and Xenopus laevis) and fish (Tetraodon nigrovirdis and Danio rerio). The sequence of the assumed 19 amino acid deletion p.Pro43_Pro61del shows a high degree of conservation in rat, mouse and opossum. The p.Pro52, p.Cys53 and p.Ala55 residues were also completely conserved in platypus (Ornithorhynchus anatinus), frog and fish.
Functional investigation of novel TBX1 variants
The mRNA expression of the wild-type construct and the two constructs incorporating mutations that changed the amino acid sequence (c.115G→A and c.129_185del57) was shown to be equal by relative quantification real-time PCR (p>0.05; data not shown). In the dual luciferase assay, the p.Gly39Ser TBX1 protein showed no significant difference in transcriptional activity from wild-type TBX1 (p=0.23; figure 1). The p.Pro43_Pro61del TBX1 protein, however, showed a significant 40% reduction in transcriptional activity compared with wild-type TBX1 (p=0.008; figure 1). Immunocytochemistry demonstrated that both the p.Gly39Ser mutated protein (not shown) and the 19 amino acid deleted protein locate to the nucleus, indicating that these mutations do not perturb protein localisation (figure 2). We next assessed the stability of the protein expressed from the c.129_185del57 TBX1 construct. There was no significant difference in the basal level of TBX1 protein expressed in cells transfected with the wild-type TBX1 or mutated TBX1 construct in three independent experiments (p=0.549). There was a significant difference in the rate at which intracellular TBX1 levels decreased after treatment with cycloheximide with the mutant protein being less stable in three independent experiments (p=0.003; figure 3).

Transcriptional activation of a luciferase reporter by wild-type (WT) and mutant TBX1. Wild type TBX1-pcDNA3 construct activates transcription, as does the G39S missense construct (c.115G→A), whereas the c.129_185del57 construct, resulting in an in-frame 19 amino acid deletion (p.Pro43_Pro61del) shows a significantly reduced transcriptional activity compared with the wild-type (40%, p=0.008). Results were normalised to Renilla luciferase for transfection efficiency and to pcDNA3 and are expressed as average values ± SD of three replicates. Results were confirmed in another three independent assays, all showing significant p values (0.002–0.02).

Subcellular localisation of TBX1 wild-type and mutant protein in transiently transfected HEK293 cells stained with 4,6-diamino-2-phenylindole (DAPI) and analysed by immunocytochemistry with anti-Tbx1 antibody.
{kind=link}
{kind=link}
{kind=link}
(A) Representative western blot showing TBX1 protein expression after 0–6 h cycloheximide treatment in HEK293 cells transfected with either the wild-type (WT) TBX1-pcDNA3 construct or the c.129_185del57 TBX1-pcDNA3 construct. The 19 amino acid deletion in the p.Pro43_Pro61del TBX1 protein results in the smaller protein size detected in these samples. Both bands were TBX1-specific (checked using an empty pcDNA3 transfected control, data not shown). The upper TBX1 band for both wild-type and p.Pro43_Pro61del samples is believed to result from phosphorylation or other post-translational modification of TBX1 as the lower band is of the expected size for wild-type TBX1 isoform C (∼56 kDa). (B) Scatter plot showing TBX1 protein expression normalised to nucleolin (loading control) for three independent experiments 0–6 h after cycloheximide treatment of cells transfected with the wild-type TBX1 construct (full squares) compared with cells transfected with the c.129_185del57 TBX1 construct (open circles). Regression analysis of the three replicates describes the slopes of the lines; −4.6% (wild-type TBX1) and −17.8% (p.Pro43_Pro61del TBX1), as significantly different (p=0.003).
Association analysis of htSNPs
The founder genotypes for all 20 TBX1 htSNPs were shown to be in Hardy–Weinberg equilibrium (p>0.01). Genotype and haplotype frequencies were in good agreement with HapMap data, for those markers where it was available. The likelihood ratio statistics and associated p values from the UNPHASED genotype analysis of the 20 TBX1 htSNPs in the TOF families and controls are shown in table 1. Three SNPs (rs739374, rs6518580 and rs5993820) showed borderline significant association (p=0.016–0.025) with TOF in 356 cases. However, this seems unlikely to reflect true association as these results do not withstand correction for multiple testing.
Association analysis of TBX1 genotypes
Discussion
We have carried out a comprehensive analysis of both common and rare TBX1 genetic variation in patients with non-syndromic TOF. Sequencing TBX1 exonic and splice site regions identified four previously unreported coding variants; three of which (c.115G→A, c.129_185del57 and c.1074G→A) were not present in 1000 control chromosomes. The c.129_185del57 variant, which deletes 19 evolutionarily conserved amino acids, was found to reduce the transcriptional activity of TBX1 by 40%. The decreased transcriptional activation observed supports our a priori hypothesis that if among the developmental processes perturbed by 22q11.2 deletion, normal cardiovascular development is particularly susceptible to differences in TBX1 activity, then hypomorphic alleles may confer risk of non-syndromic CHD. As the p.Pro43_Pro61 deletion is outside the DNA binding domain, which extends from aa119 to aa297, we next assessed whether subcellular localisation or protein stability were affected by the mutation. We found that the mutant protein localised appropriately to the nucleus but that it was less stable than wild-type protein. Thus a mechanism by which it leads to decreased transcriptional activity is that TBX1 protein levels are diminished, though this may not be the sole mechanism—for example, the deletion may also affect dimerisation or interaction with other partners. It is interesting that in addition to TOF this child also had a right-sided aortic arch, an abnormality reported in Tbx1 mutant mice.13 The c.115G→A, p.Gly39Ser variant did not affect transcriptional activity in the in vitro assay; however, this assay does not fully recapitulate the complex interactions occurring in the developing embryo. Transcription factors act on target genes in concert and as more becomes known about TBX1 protein interactions the effect of this mutation on protein interactions should be investigated. As yet, little is known about Tbx1 protein interactions but Tbx1 has recently been shown to interact with Srf (serum response factor), a regulator of muscle differentiation,22 and with SMAD1, affecting BMP signalling in a transcription independent manner.23
Although TBX1 mutations had been identified in studies of patients who have clinical features of chromosome 22q11.2 deletion syndrome, including CHD, but who are not deleted for the region,16–19 early studies of TBX1 in non-syndromic TOF were small and failed to identify mutations. A substantially larger recent study of 191 patients with TOF who did not have a chromosomal abnormality or recognised syndrome identified two novel TBX1 variants.3 One of these p.Pro290Ser, not present in 174 control individuals and inherited from a phenotypically normal father, did not affect transcriptional activity in vitro. The second, a 30 bp insertion, leading to expansion of a polyalanine tract, decreased transcriptional activity in vitro; further studies showed aggregation of the expressed mutant protein in the cytoplasm. Taking the two studies together it is evident that TBX1 coding sequence mutations do occur in patients with non-syndromic TOF but account for only about 1% of cases.
A recent study has demonstrated a non-linear relationship between Tbx1 mRNA dosage and the 22q11.2 deletion syndrome phenotype, including cardiac OFT defects, in the mouse.24 The penetrance of cardiac OFT defects was shown to sharply increase at mRNA concentrations of less than about 20% of normal in that study; the equivalent threshold for TBX1 activity in man is unknown, although our study suggests that human OFT development is sensitive to smaller decreases in TBX1 activity. In vitro studies of three TBX1 missense changes identified in patients with the 22q11.2 phenotype found increased transcriptional activity.19 Similarly, investigation of a 5′ untranslated change, -39C→T, identified in patients with the 22q11.2 phenotype showed this to be associated with increased transcript levels.16 25 Thus human OFT development appears to be sensitive to decreased activity and also to increased TBX1 activity.
Systematic study of all common variation in the TBX1 region in 356 TOF cases, together with available parents and unrelated controls, provided no support for the hypothesis that common SNPs in this region have sizeable effects (ie, conferring ORs >2.0) on the risk of TOF, although the possibility smaller effects of common variants cannot be ruled out. No previous studies have investigated the potential contribution of common TBX1 genetic variants in TOF. For common conditions such as coronary disease, the contribution of SNPs to risk, discovered by genome-wide association studies, has been small, with ORs generally <1.3. However, some rarer diseases have been shown to be influenced to a greater extent by common variants. For example, Crohn's disease has a population prevalence approximately equal to that of congenital heart disease. SNPs in the Nod2/CARD15 gene with minor allele frequency ∼0.05 are associated with Crohn's disease risks of 2.2–4.0 to heterozygotes and around 17.0 to homozygotes26; this study had >80% power at p<10-5 to detect CHD risks of this magnitude associated with SNPs of similar frequency. Previous studies suggesting an oligogenic aetiology for TOF increased the a priori likelihood that effects of this size might be present.6 We had low power to detect common variants associated with relative risks <2.0 and larger studies will be required to conclusively establish the presence or absence of such smaller effects at TBX1. More generally, genome-wide association studies in large numbers of patients and family-based or unrelated controls will be required to resolve whether common genetic variation importantly influences non-syndromic CHD susceptibility.
In summary, this study is the first comprehensive investigation of common and rare TBX1 genetic variants in non-syndromic TOF cases. It has identified a rare novel functional genetic variant that is a likely susceptibility factor to TOF. However, as the study indicates that mutations in TBX1 account for at most a few per cent of the susceptibility to non-syndromic TOF it does not support the introduction of TBX1 mutation analysis to clinical practice in this patient group; systematic large-scale surveys of other CHD candidate genes will be necessary to identify the genetic risk factors accounting for the bulk of the heritability seen in epidemiological studies.
Acknowledgments
The principal acknowledgement is to the patients and families who participated in this study. We thank study nurses K Ashcroft, L Sneddon, C Roberts, J Peters, H Walker and R Hussein for technical support.
References
Footnotes
Funding This work was supported by the British Heart Foundation (RG/02/014); European Community's Sixth Framework Programme contract ('HeartRepair') (LSHM-CT-2005-018630); and the Federated Foundation; BK holds a British Heart Foundation Chair; HRG was supported by a Medical Research Council PhD studentship.
Competing interests None.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.