Article Text

Abstract
Background In patients with acute myocardial infarction, restoration of coronary flow by primary coronary intervention (PCI) can lead to profound ischaemia-reperfusion injury with detrimental effects on myocardial salvage. Non-invasive assessment of interstitial myocardial haemorrhage by T2* cardiac MRI (T2*-CMR) provides a novel and specific biomarker of severe reperfusion injury which may be of prognostic value.
Objective To characterise the determinants of acute ischaemia-reperfusion injury following ST elevation myocardial infarction (STEMI) using CMR.
Methods and results Fifty patients with acute STEMI who had been successfully treated by PCI were studied. T2*-CMR was used to identify the presence of reperfusion haemorrhage and contrast enhancement was used to measure microvascular obstruction (MVO) and infarct size. Haemorrhagic ischaemia-reperfusion injury was present in 29 patients (58%) following PCI and occurred despite rapid revascularisation (mean 4.2±3.3 h). Haemorrhage was only present when the infarct involved at least 80% (mean±SD 91±5.3%) of the left ventricular wall thickness. There was a strong association between the extent of MVO and reperfusion haemorrhage (r2=0.87, p<0.001). Transmural infarcts (n=43) showed significantly impaired systolic wall thickening at the infarct mid point when reperfusion haemorrhage was present (21.5±16.7% vs 3.7±12.9%), p<0.0001) compared with non-haemorrhagic infarcts.
Conclusions Severe reperfusion injury may occur when there is near-transmural myocardial necrosis despite early and successful revascularisation. Reperfusion haemorrhage is closely associated with the development of MVO. These findings indicate that, once advanced necrosis has developed, the potential for severe myocardial reperfusion injury is significantly enhanced.
- MRI
- myocardial infarction
- myocardial reperfusion injury
- no-Reflow phenomenon
- Angioplasty, transluminal, percutaneous coronary
- MRI
- acute coronary syndrome
- reperfusion
Statistics from Altmetric.com
- MRI
- myocardial infarction
- myocardial reperfusion injury
- no-Reflow phenomenon
- Angioplasty, transluminal, percutaneous coronary
- MRI
- acute coronary syndrome
- reperfusion
Early restoration of myocardial perfusion is the most important goal of treating patients with acute ST elevation myocardial infarction (STEMI).1 However, despite re-establishing patency of the infarct-related coronary artery, achieving optimal tissue perfusion and myocyte salvage in all patients has remained elusive.2 The process of restoring blood flow to ischaemic myocardium can induce damage itself, and this phenomenon—termed ischaemia-reperfusion injury—can paradoxically reduce the beneficial effects of myocardial reperfusion.3 This process results in the death of cardiac myocytes that were viable immediately prior to myocardial reperfusion and may be responsible for up to half the final infarct size.4
Determining the conditions under which reperfusion injury occurs in clinical practice would provide a rationale for treating or avoiding this therapeutic complication. In recent years there has been increasing interest in cardiac MRI for assessing myocardial ischaemic injury.5 T2-weighted imaging of myocardial oedema has been shown to be an accurate method of retrospectively determining the extent of ischaemic tissue in both animal models and in clinical practice after primary coronary intervention (PCI).6 7 Following revacularisation, myocardial oedema is present in both non-reperfused8 and reperfused infarcts7; however interstitial myocardial haemorrhage is only observed following coronary reperfusion and serves as a specific marker of severe ischaemia-reperfusion injury.9–11 Furthermore, the presence of interstitial haemorrhage is strongly associated with microvascular obstruction (MVO) which is known to rapidly worsen following reperfusion.12
Haemorrhage contains paramagnetic blood degradation products which have characteristic effects on the magnetic field at the microscopic level and can be readily identified with T2*-weighted cardiac MRI (T2*-CMR).13 Mapping the distribution of these blood products provides the only non-invasive method for identifying infarctions where severe reperfusion injury has occurred. We used this approach to assess the conditions which predispose to the development of severe reperfusion injury in patients with acute STEMI treated with PCI.
Methods
Patients
The study was approved by the hospital research ethics committee and all patients gave written informed consent. Fifty patients (48 men, 2 women; age range 29–74 years; mean age 54.8 years) who had undergone PCI within the previous 7 days were prospectively investigated. Fifteen of these subjects had participated in previously published work.13 14 The inclusion criteria were men and women aged 18−85 years with an ECG diagnosis of acute STEMI who were treated with PCI. Exclusion criteria were contraindications to CMR, previous myocardial infarction or heart failure, clinical instability, significant arrhythmias, pregnancy or lactation. The diagnosis of STEMI required ST elevation of ≥1 mm in two contiguous ECG leads and chest pain onset within 12 h of presentation.
Coronary intervention
Prior to PCI the patients received aspirin and clopidogrel, and intravenous heparin was administered as a bolus with the aim of achieving an activated clotting time of >250 s. Patients underwent left heart catheterisation using either transfemoral or transradial approaches. Selective coronary catheterisation was used to identify and treat the infarct-related artery and all patients received either a bare metal or drug-eluting stent. Non-culprit atherosclerotic vessels were not treated. In total, 44 patients (88%) received a glycoprotein IIb/IIIa inhibitor periprocedurally.
Angiographic analysis
Flow in the infarct-related artery was graded using the Thrombolysis in Myocardial Infarction trial (TIMI) criteria prior to and after the intervention.
Image acquisition
The CMR studies were performed on a 1.5T Philips Achieva system (Best, The Netherlands). The maximum gradient strength was 33 mT/m and the maximum slew rate was 160 mT/m/ms. A five-element cardiac phased-array coil was used for signal reception.
Scout images were obtained and used to plan an axial stack of cine balanced-steady state free precession (b-SSFP) images in the left ventricular short axis plane from base to apex using the following parameters: voxel size 2.0 × 2.2 × 8 mm, flip angle 60°, slice thickness 8 mm with a 2 mm gap, TE 1.5 ms, TR 3.0 ms and 20 cardiac phases. Myocardial oedema was imaged with a navigator-gated black blood T2-weighted turbo spin echo sequence with spectrally selective inversion recovery (SPIR) fat suppression using the following parameters: voxel size 1.9 × 2.6 × 10 mm, flip angle 90°, slice thickness 10 mm, TE 100 ms, TR 2 R–R intervals. Myocardial haemorrhage was imaged with a navigator-gated black blood gradient multiecho T2* sequence using the following parameters: voxel size 1.7 × 2.8 × 10 mm, flip angle 20°, slice thickness 10 mm, TR 17 ms, 7 echoes, TE 2.3–16.1 ms, ΔTE 2.3 ms. A single short-axis image plane was chosen at the level of maximal oedema.
An intravenous bolus of gadolinium dimeglumine (Magnevist; Bayer Schering Pharma, Berlin, Germany) was administered at a dose of 0.2 mmol/kg via a power injector (Spectris Solaris; Medrad, Indianola, Pennsylvania, USA). Early enhancement imaging was performed with a single breath hold 3D inversion recovery sequence within 1 min of contrast injection using the following parameters: voxel size 1.4 × 2.1 × 8 mm, flip angle 15°, slice thickness 8 mm, TE 1.4 ms, TR 4.3 ms. Late enhancement imaging was performed after a 10 min delay with a 2D inversion recovery sequence using the following parameters: 1.5 × 1.7 × 8 mm, flip angle 15°, slice thickness 8 mm, TE 1.4 ms, TR 4.3 ms. The inversion time was adjusted to null the signal from normal myocardium using the Look-Locker method.
Image analysis
Segmentation of the left ventricular cavity and wall from the short axis b-SSFP cine images was performed using CMRtools (Cardiovascular Imaging Solutions, London, UK). Measurements were made by two experienced observers and the mean value obtained. Left ventricular mass, end diastolic volume and end systolic volume were calculated and indexed to body surface area. End diastolic wall thickness and fractional wall thickening were measured at the mid point of the infarction. The regions of late enhancement and MVO were quantified using an automated algorithm (Segment 1.8, Medviso, Sweden) with a section-specific signal intensity threshold and level-set boundary detection to identify the interfaces between different regions of signal intensity.15 A single section in the same plane as the short axis T2* image was used for comparative analysis and infarct transmurality was measured along 40 radial cords.
A pixel-by-pixel analysis was performed of the multiecho T2*-weighted images using Matlab 2008b (Mathworks, Natick, Massachusetts, USA) with fitting to a simple monoexponential decay. The T2* decay constant was measured in both haemorrhagic and non-haemorrhagic myocardium. The proportion of pixels within infarcted myocardium with a T2* value of <20 ms (retrospectively determined as 2SD below the mean of remote myocardium in all subjects) was measured and expressed as a percentage of the left ventricular area of that section. Reperfusion haemorrhage was considered to be present if the area of abnormal T2* within the confines of the infarct constituted at least 1% of the left ventricular area in that section. Susceptibility artefact adjacent to the inferolateral left ventricular epicardium was manually excluded if present. Haemorrhage was considered to be present on T2-weighted imaging if there was a hypointense area within the area at risk with a mean signal intensity 2SD below the signal intensity of myocardial oedema constituting at least 1% of the left ventricular area in that section.16 The proportion of area at risk on T2-weighted imaging which did not show late enhancement was used to calculate the myocardial salvage index.
Statistical analysis
All continuous data are reported as mean±1SD. Comparison of continuous variables between groups was performed with the Mann–Whitney U test. Categorical variables were compared with the χ2 test. The association between variables was assessed by linear least squares regression. In each case a p value of <0.05 was considered statistically significant. Univariate and multivariate regression analysis was used to assess the predictive value of CMR findings for post-PCI ejection fraction. Variables were retained in the model if they contributed to the explanatory power of the regression equation with a p value <0.10.
Results
Patient characteristics
On the basis of T2*-CMR, 29 patients had haemorrhagic infarctions and 21 patients had non-haemorrhagic infarctions. The baseline parameters are shown in table 1. For all patients the mean time from symptom onset to reperfusion was 3.9±2.8 h (range 1–12). The baseline CMR was performed at a mean of 3.1±2.0 days (range 1–7) after PCI. All subjects demonstrated myocardial oedema within the vascular territory of the culprit artery and this was invariably associated with a regional wall motion abnormality. On late enhancement images all patients showed a single contiguous zone of myocardial necrosis. Late enhancement was located within the zone of oedema in each case. The mean infarct size for all patients was 18.9±9.3% (range 6–41%) with a mean salvage index of 45.2±21.6% (range 0–85%).
Baseline characteristics of patients undergoing primary coronary intervention
Microvascular obstruction and reperfusion injury
MVO was present in 29 patients (58%). The mean area of early MVO was 19.0±11.0% (range 5.4–40.5%) and late MVO was 6.6±4.8% (range 1.0–18.5%). Small regions of MVO were typically confined to the subendocardium but, in more extensive infarcts, only the subepicardium was spared. Focal myocardial haemorrhage was identified with T2* imaging in all patients with MVO and the regions of haemorrhage and MVO were co-localised (figure 1). The mean area of haemorrhage was 7.7±6.2% (range 1.0–25.4%) and was present in infarcts with a cross-sectional area between 17% and 57% of the left ventricle. The area of haemorrhage showed a strong correlation to the corresponding area of late MVO (r2=0.87, p<0.001) but a weaker correlation with the larger and more variable area of early MVO (r2=0.30, p<0.003).
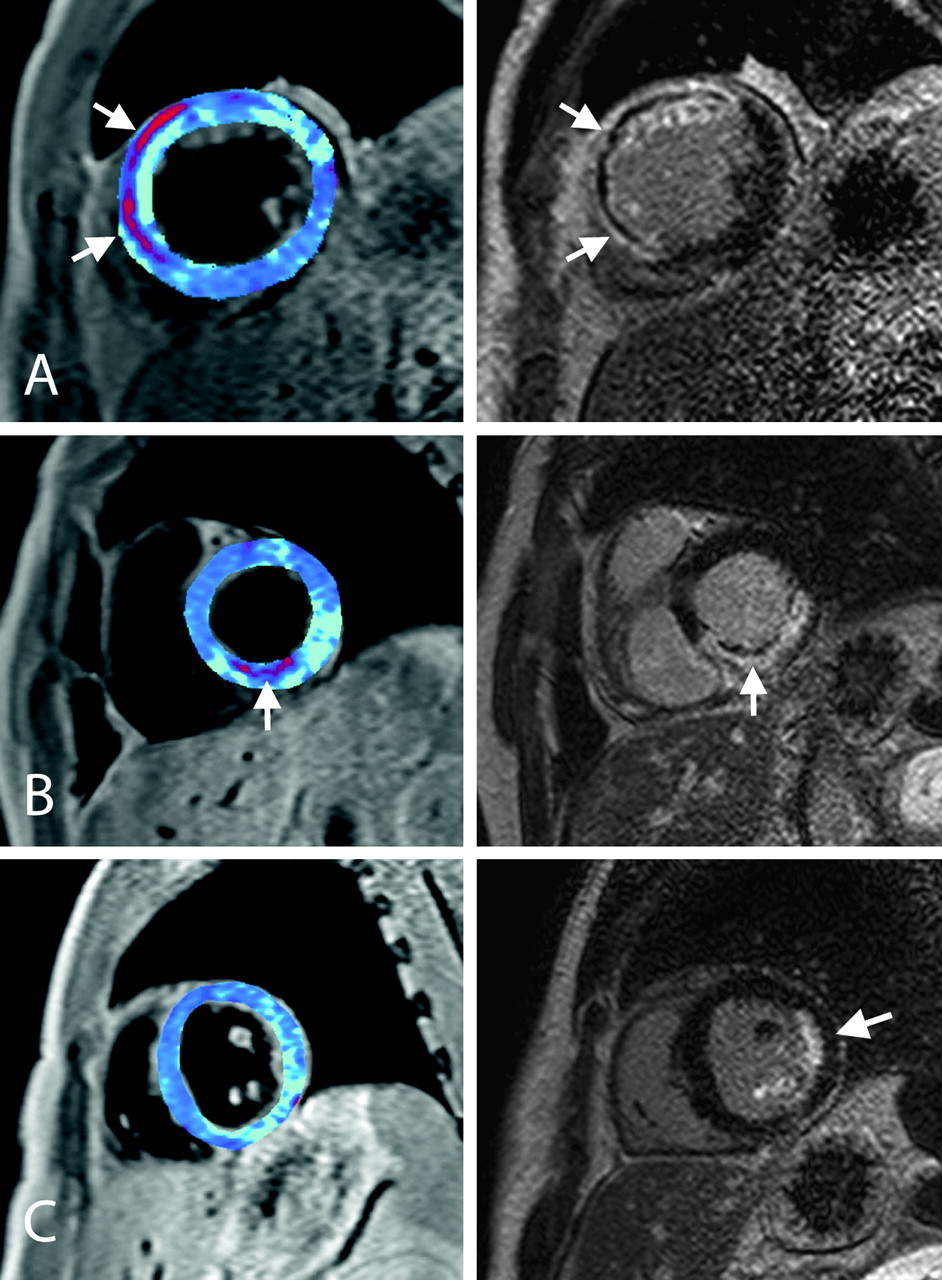
Cardiac MRI of acute reperfusion injury. Images from three patients (A, B and C) with acute ST elevation myocardial infarction obtained within 48 h of primary coronary intervention. The left panels show the T2* maps with areas of haemorrhage depicted in red. The right panels show the delayed contrast-enhanced images. (A) A left anterior descending coronary artery occlusion showing a close correspondence between reperfusion haemorrhage and microvascular obstruction (MVO) (arrows) in the anterior wall. (B) A right coronary artery occlusion demonstrating subendocardial haemorrhage and MVO in the inferior wall (arrow). (C) An obtuse marginal artery occlusion resulting in a partial thickness infarct of the lateral wall (arrow) and posterior papillary muscle; however, neither haemorrhage nor MVO is present in this case.
T2-weighted imaging versus T2* mapping
T2* mapping had a sensitivity of 100% for detecting the presence of MVO in an equivalent section. Susceptibility artefact, particularly for lateral wall infarcts, resulted in a specificity of 90%. The mean T2* within haemorrhagic infarctions was 15.4±5.7 ms compared with 47.2±13.8 ms in non-haemorrhagic myocardium. T2-weighted imaging of oedema had a sensitivity of 82% for detecting MVO, and an example of a false negative study is shown in figure 2. The specificity for T2-weighted imaging was also 90%.

Reperfusion haemorrhage only apparent on T2* imaging. Cardiac MRI images obtained from a patient 3 days after primary coronary intervention to a left anterior descending coronary artery. (A) T2-SPIR (spectrally selective inversion recovery) imaging shows uniform acute myocardial oedema in the septum (arrowheads). (B) A T2* map shows the areas of reperfusion haemorrhage in red (arrow). This closely corresponds to the region of persistent microvascular obstruction on the late enhancement image (C, arrow) which is surrounded by enhancing necrosis.
Characteristics of haemorrhagic infarctions
Haemorrhagic infarctions were observed in each of the main coronary artery territories. In haemorrhagic infarcts, all 29 patients had either TIMI grade 0 or I prior to intervention. Following PCI, 27 patients (93%) had grade III flow and 2 patients (7%) had grade II flow. All patients were treated with aspirin and heparin periprocedurally, and there was no significant difference in the rates of glycoprotein IIb/IIIa antagonist usage between haemorrhagic and non-haemorrhagic infarcts. Cardiac enzymes (troponin I and creatinine kinase) were significantly greater in the haemorrhagic infarction group (p<0.05). No significant difference in pain-to-balloon time was identified between haemorrhagic and non-haemorrhagic infarcts, with reperfusion haemorrhage detected in subjects who received PCI within the first few hours after the onset of chest pain.
The imaging findings are summarised in table 2. Infarct volume was significantly greater when haemorrhage was present (23.8±8.2% vs 12.0±5.9%, p<0.0001). The left ventricular ejection fraction (LVEF) was lower in the haemorrhage group (51±11% vs 59±7%, p<0.01). Using univariate analysis, the area of reperfusion haemorrhage showed a strong association with LVEF (β=−0.56, p<0.0001). After multivariate analysis, infarct volume remained the only independent predictor of LVEF (β=−0.60, p<0.01).
Cardiac MRI parameters following primary coronary intervention
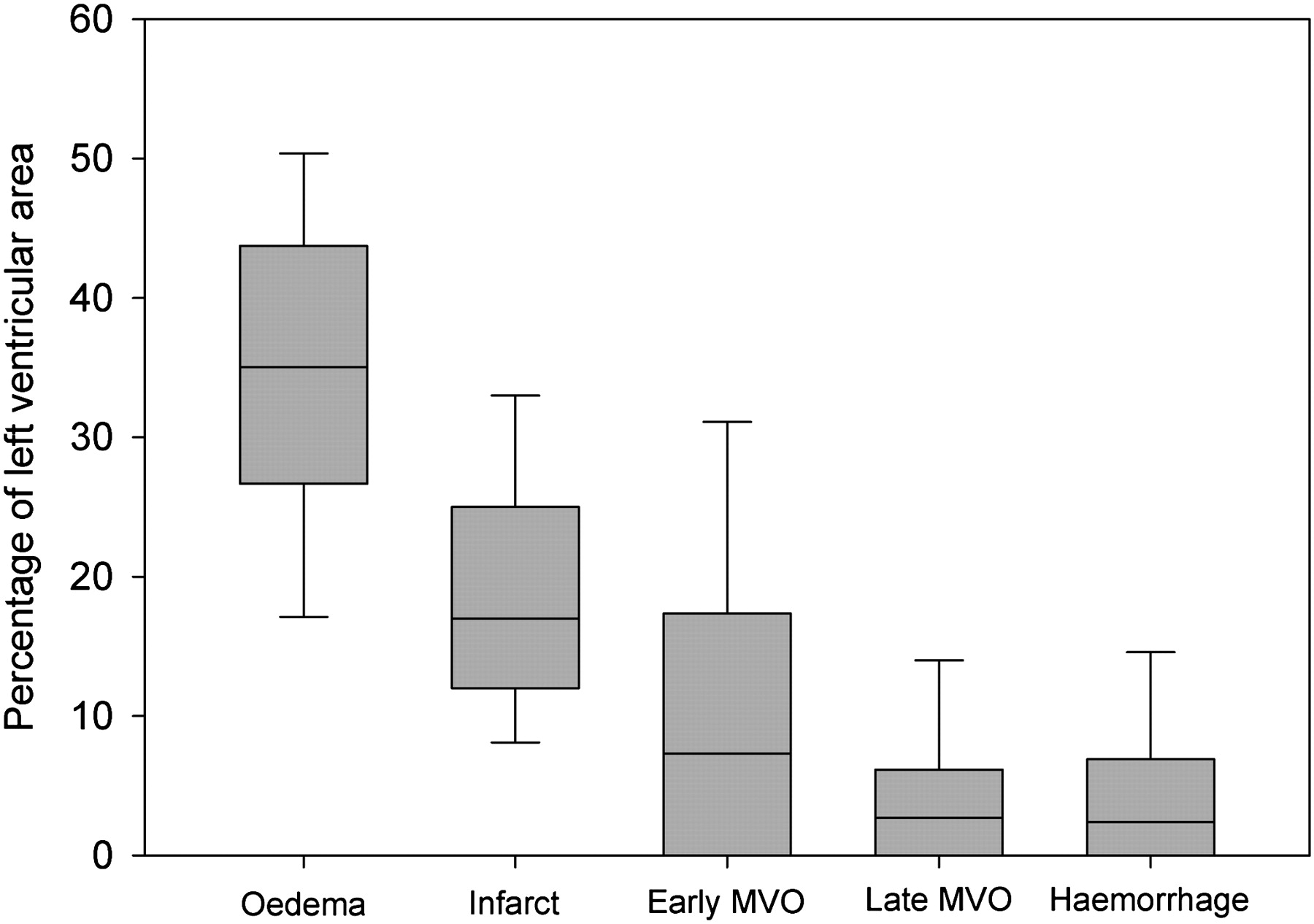
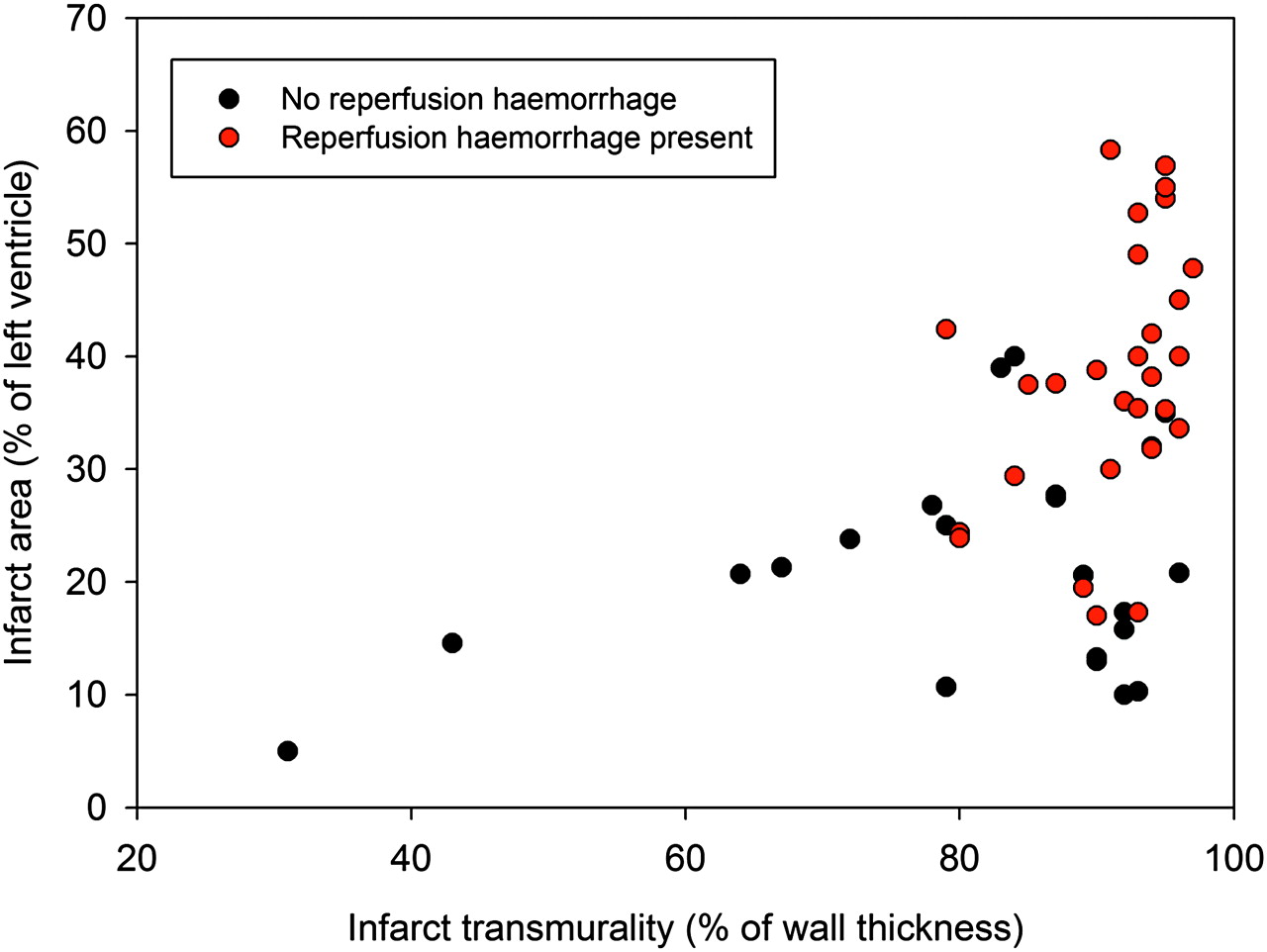
Excluding the seven patients with partial thickness infarcts (necrosis <80% of left ventricular wall), fractional wall thickening at the infarct mid point showed significantly greater impairment when reperfusion haemorrhage was present (21.5±16.7% vs 3.7±12.9%, p<0.001). Haemorrhagic infarction was associated with significantly greater end diastolic volume index (81.0±21.4 ml/m2 vs 67.1±11.4 ml/m2, p<0.001) and end systolic volume index (41.1±17.1 ml/m2 vs 28.3±7.9 ml/m2, p<0.001). No significant difference was found between end diastolic wall thickness at the infarct mid point, although left ventricular mass was elevated in haemorrhagic infarctions (84±16 g/m2 vs 73±11 g/m2, p<0.01). The relationship between cross-sectional area of the infarct and infarct transmural thickness for both haemorrhagic and non-haemorrhagic infarcts is shown in figure 3. A plot of the relative extent of myocardial oedema, infarct, early MVO, late MVO and haemorrhage is shown in figure 4.

Scatterplot of infarct transmurality and cross-sectional infarct area. Severe reperfusion injury (red points) is more prevalent in larger infarcts where necrosis is almost transmural.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Box plot of the relative areas of ischaemia-reperfusion injury. Characteristics of the areas of infarct tissue determined with cardiac MRI measuring oedema with T2-SPIR, early microvascular obstruction (MVO) with early enhancement, late MVO and infarct size with delayed enhancement, and haemorrhage with T2* mapping (boxes show the median value and are bounded by the 25th and 75th centiles, error bars are at the 10th and 90th centiles).
Discussion
Ischaemia-reperfusion injury is known to have a major influence on myocyte salvage and final infarct size. Our study has used a novel imaging technique to quantify the presence of myocardial haemorrhage which is indicative of severe reperfusion injury in post-STEMI patients. We observed that reperfusion injury is rarely present in smaller subendocardial infarcts but is a frequent complication in large infarcts where necrosis is almost transmural, which can occur even in rapidly reperfused infarcts. These findings indicate that, as necrosis becomes established throughout the ischaemic bed, the potential for an adverse response to reperfusion is significantly enhanced.
The concept of lethal reperfusion injury as an independent mediator of myocyte death, as distinct from an exacerbation of ischaemic injury, is controversial.3 The difficulty lies in determining the progress of necrosis during the transition from ischaemia to reperfusion.4 Although T2-weighted imaging allows determination of the ischaemic area at risk, it is not possible to retrospectively determine the effect of reperfusion on oedema and necrosis as both of these features are also observed in non-reperfused infarcts. While ischaemia-reperfusion injury has an effect on vulnerable myocytes throughout the vascular bed of the culprit artery,17 the development of haemorrhage appears to be a specific finding that occurs only after flow has been restored to severely ischaemic myocardium.9–11 The technique of T2* imaging used in our study has been validated in both preclinical and ex vivo studies as an effective method for quantifying reperfusion haemorrhage and comparing its distribution with other measures of ischaemic damage.18 19
In our study, reperfusion injury was not restricted to late presentations and in some cases haemorrhage was present despite early and successful coronary intervention. We observed severe reperfusion injury in infarcts with poor myocardial salvage, especially in the left anterior descending (LAD) coronary artery territory, and did not find a strong association with pain-to-balloon time. This may reflect the wide interindividual variation in salvage rates achieved with PCI.20 Indeed, our findings indicate that, once necrosis has become established within the ischaemic bed, reperfusion haemorrhage may occur rapidly and extensively within the infarct. We also observed that haemorrhage occurs in the subendocardium and mid wall, which is consistent with the secondary wavefront of reperfusion injury seen in animals.21 We found that reperfusion haemorrhage consistently coincided with the area of irreversible microcirculatory damage (MVO). The extent of MVO increases significantly even after the restoration of coronary flow.12 The mechanism for this is unknown, although vascular microplugging by platelets and neutrophils, myocyte cell swelling and free radical-mediated endothelial injury have all been proposed.22 In animal studies the advance of reperfusion haemorrhage lags behind the expansion of MVO,21 but the relationship between these phenomena in clinical practice is unclear, especially as patients are routinely heparinised and treated with antiplatelet drugs. It has been proposed that, if the expansion of MVO can be closely related to progressive haemorrhage within the myocardium, it might be considered a reperfusion injury which could be potentially avoided or treated.23 In our study the highly consistent association of reperfusion haemorrhage with MVO throughout the first 7 days following PCI indicates that haemorrhage expansion has a potential role in the evolution of MVO.
The presence of MVO is associated with adverse left ventricular remodelling even after controlling for infarct size.24 If reperfusion injury is a contributory factor in the development of MVO, it raises the question of whether attempting reperfusion of a completed infarct may worsen the patient's prognosis. Although experimental studies have shown a reduction in infarct expansion and left ventricular dilation even when late reperfusion has been initiated in completed infarcts,25 26 clinical trials have failed to show that the ‘open-artery hypothesis’ leads to improved clinical outcomes.27 28 Studies of the prognostic significance of reperfusion haemorrhage as an independent predictor of remodelling have been inconclusive although, until recently, investigators have relied on detecting signal voids on T2-weighted imaging within the area of myocardial oedema to infer the presence of haemorrhage.16 29 In the study of reperfusion injury, T2*-CMR is advantageous as it is more sensitive to the susceptibility effects of subacute haemorrhage than spin echo imaging and does not rely on the presence of myocardial oedema.30 Further research will be needed to determine the independent predictive contribution of reperfusion haemorrhage in ventricular function and remodelling. We identified infarct volume as the only independent predictor of baseline ejection fraction, and this reflected the strong colinearity of two independent variables (haemorrhage and MVO) in the regression model. As well as risk stratification, T2*-CMR may also prove useful in the investigation of cardioprotective therapies by providing a specific biomarker of reperfusion-induced tissue injury.
A limitation of this study was that no histopathological correlation with imaging findings was possible as all of the patients survived, and the validity of our observations is based on published animal and ex vivo studies. The sizing of haemorrhage was based on a T2* threshold derived from comparison with remote myocardium; however, the maturation of blood products in the first week following PCI may affect the degree of paramagnetic susceptibility. T2* imaging was also only performed at one level in the heart. In common with previous studies, we observed that the extent of apparent MVO decreases with time following gadolinium administration.23 Our findings demonstrated a stronger correlation between haemorrhage and the extent of MVO on delayed sequences, which suggests that the haemorrhagic infarct core has the most severe impairment of microvascular flow. However, the optimal methods for sizing MVO have not been determined.
Conclusion
Severe reperfusion injury may occur when there is near transmural myocardial necrosis despite early and successful revascularisation. Reperfusion haemorrhage is closely associated with the development of MVO. These findings indicate that, once advanced necrosis has developed, the potential for severe myocardial reperfusion injury is significantly enhanced.
References
Footnotes
Funding Medical Research Council, UK.
Competing interests None.
Ethics approval This study was conducted with the approval of the Hammersmith Hospital research ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.