Article Text

Abstract
Mutations involving cardiac ion channels result in abnormal action potential formation or propagation, leading to cardiac arrhythmias. Despite the large impact on society of sudden cardiac death resulting from such arrhythmias, understanding of the underlying cellular mechanism is poor and clinical risk stratification and treatment consequently limited. Basic research using molecular techniques, as well as animal models, has proved extremely useful in improving our knowledge of inherited arrhythmogenic syndromes. This offers the practitioner tools to accurately diagnose rare disorders and provides novel markers for risk assessment and a basis for new strategies of treatment.
- Channelopathy
- sudden cardiac death
- Brugada
- ventricular tachycardia
- sudden adult death syndrome
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
Ion channels are pore-forming proteins that provide pathways for the controlled trans-membrane movement of ions. This is critical for a range of physiological processes including action potential (AP) generation and propagation, resulting in the release of intracellular Ca2+ stores triggering mechanical activity. Abnormalities in cardiac ion channel function or in their associated regulatory proteins may lead to arrhythmias and sudden cardiac death (SCD). SCD poses a major medical challenge and significant public health burden, accounting for over 300 000 deaths per year in the USA,1 and up to 70 000 deaths per year in the UK,2 with survival rates of only 2%. In the majority of cases, the arrhythmias are a manifestation of underlying ischaemic heart disease; however, autopsy fails to reveal a cause in up to 40% of SCD patients.3
Despite the high prevalence and large impact on society of cardiac arrhythmias, our understanding of the cellular and molecular mechanisms governing the initiation, maintenance and propagation of arrhythmias remains limited. Consequently, current risk stratification of patients and families with these conditions is inadequate and the mainstay of treatment is often restricted to implantable cardioverter defibrillator implantation. Although new techniques are being developed to investigate the mechanisms that predispose to SCD, invasive studies in humans are limited. Therefore, basic research using molecular techniques as well as animal models is essential in improving our understanding of the mechanisms of arrhythmogenesis at the cellular level. This review forms the third paper in a series of inherited channelopathy reviews published in this journal, with the earlier papers discussing the impact of pathophysiology4 and the role of the Sudden Adult Death Syndrome clinic5a in the management of ion channel disease. The current review will focus on the role of basic science in investigating primary electrical diseases of the heart as a paradigm for cardiac arrhythmias, concentrating on Brugada syndrome (BrS), long QT syndrome (LQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT).
Mechanisms of ventricular arrhythmogenesis
Normal cardiac excitability results from a balance of depolarising and repolarising ionic currents. Each ionic current can be distinguished by its ionic selectivity and time course, which are properties that are conferred by specific ion channels (for more details see review by Behr et al5b). Mutations in any of the genes involved in regulation of cardiac ion channels may potentially result in arrhythmias and may be classified as arising from either abnormal AP formation or abnormal AP propagation (figure 1).

Mechanisms contributing to arrhythmogenesis. (A) Triggered activity, the upper trace shows an early after-depolarisation (EAD) occurring during repolarisation. The lower trace shows a delayed after-depolarisation (DAD) occurring following repolarisation. Dotted lines indicate the generation of an extra action potential with a sufficiently sized after-depolarisation. (B) Re-entrant circuit, the upper diagram shows a normal pattern of excitation with impulse propagating equally at each bifurcation and extinguishing in the middle. The lower diagram shows an area of slowed conduction velocity in one of the branches. The normal impulse shown by the solid arrow does not get extinguished as indicated by the delayed dotted arrow, allowing it to excite the myocardium. The delayed impulse is then able to circulate around the junctions resulting in inappropriate excitation of the rest of the myocardium. (C) Spatial heterogeneities in the form of transmural gradients. The epicardial action potential duration is much shorter than the endocardial potentially leading to re-excitation. (D) Temporal heterogeneities in the form of discordant action potential duration alternans. Regions which alternate out of phase generate a line of block called the nodal line between them. This has the potential to act as a focus for re-entrant circuits following the addition of a triggered beat.
Abnormal AP formation
Abnormal AP formation may occur as a result of enhanced automaticity, parasystole or triggered activity. Enhanced automaticity describes the accelerated depolarisation of pacemaker tissue that might occur as a result of increased sympathetic activity, hypokalaemia or drugs such as digitalis. Parasystole refers to the parallel activation of two of more pacemaker regions, which may override their usual suppression through protection by a region of conduction block. Both are rare causes of arrhythmias.6
Triggered activity describes the generation of extra systoles produced in regions other than the primary cardiac pacemaker (figure 1A). These are usually produced by after-depolarisations. These are oscillations of membrane potential that are a function of the preceding AP and which may generate a premature AP, and if of sufficient magnitude locally to cause a propagated wave of depolarisation can produce a triggered beat. Early after-depolarisations (EADs) occur during repolarisation of the AP while delayed after-depolarisations (DADs) do not occur until repolarisation is complete or nearly complete. EADs occur when a prolonged AP duration (APD) allows time for L-type Ca2+ channels to recover from inactivation while the membrane is still depolarised. When sufficient recovery has occurred, inward ICa-L further depolarises the membrane and initiates the after-depolarisation, thereby exerting a positive feedback effect.7 DADs are caused by enhanced Ca2+ release from the sarcoplasmic reticulum (SR) due to either to activation of the Na+/Ca2+ exchanger or Ca2+ activated Cl− current. Thus, DADs tend to occur under conditions of Ca2+ overload such as during digitalis toxicity and CPVT.8
Abnormal AP propagation: mechanisms of re-entry
The mammalian ventricular myocardium normally has a highly regulated sequence of AP activation. Abnormal AP propagation can occur when the depolarisation wavefront fails to completely extinguish after normal activation, and can therefore lead to the re-excitation of regions that have recovered excitability. This circulating wave results in the spread of depolarisation, termed re-entry (figure 1B).
There are three principal requirements for the initiation of re-entrant arrhythmias:
an obstacle around which the AP is able to circulate
a conduction velocity sufficiently slow such that each region recovers excitability before the wave returns
an existence of unidirectional conduction block, preventing the wave from self-extinguishing.
Such criteria have been demonstrated in numerous previous experiments, the earliest of which used an anatomical block,9 as may be seen in patients with ventricular scarring, usually from old myocardial infarction, but potentially also from cardiomyopathy and infiltrative disease. Mapping studies have shown the development of a line of functional unilateral block, which prevents the wave from self-extinguishing. The conduction velocity is slowed so that each region recovers excitability before the wave returns.10 Re-entry can also occur in the absence of scar, if there are regional heterogeneities in membrane excitability. Unilateral block can result if there are spatial gradients in the time taken between stimulation and depolarisation (latency) or in the time taken between depolarisation and recovery from the ventricular effective refractory period (VERP).
Spatial electrophysiological heterogeneity
The normal mammalian ventricular myocardium similarly has a highly regulated sequence of repolarisation, proceeding from epicardium to endocardium and from base to apex. The gradients that are observed in the normal heart may serve to protect the normal sequence of electro-mechanical activation and enable the correctly timed mechanical contractile activity that enables coordinated mechanical activation and relaxation of the heart. These spatial differences are determined predominantly by regional differences in repolarising K+ channels, including variations in channel density, kinetics and cycling between membrane and cytoplasm. Disturbances in these gradients may allow depolarised regions to re-excite already repolarised areas and serve as substrates for re-entry. Transmural gradients have been suggested to be particularly important as differences in repolarisation are exerted across a relatively short distance, with alterations in transmural gradients correlated with arrhythmogenic tendencies in a number of both pharmacological canine and genetic murine models for LQTS and BrS (figure 1C). As well as transmural gradients, heterogeneities within the ventricle from base to apex have been demonstrated in a number of animal models. Clinical increases in this dispersion have been associated with arrhythmogenesis in cardiomyopathies and have been linked with an increased incidence of T wave alternans and ventricular tachycardia (VT).11 Data from the canine right ventricular (RV) wedge preparation12 and the Scn5a+/− mouse model13 implicate re-entry as a result of epicardial dispersion of repolarisation as the trigger for ventricular arrhythmia. Last, differences in APDs between the left ventricle and RV may play a role in arrhythmogenesis in BrS, as demonstrated by electrocardiographic ST elevation in the right precordial leads, RV delay and changes specific to RV epicardial AP waveforms in BrS patients.14
Temporal electrophysiological heterogeneity
As well as spatial heterogeneities, there may also be temporal beat-to-beat variations in the AP amplitude or duration, a phenomenon known as alternans (figure 1D). This has been associated with arrhythmogenesis in both clinical15 and experimental16 studies. Several hypotheses exist for the cellular mechanism for the AP alternans itself; although there is a likely role for SR Ca2+ cycling, APD restitution is also a key. The latter specifically concerns the attenuation of APD that occurs in response to faster heart rates and is thought to be an adaptive mechanism for preserving diastolic interval. At a given cycle length, APD prolongation during one beat is necessarily followed by a short diastolic interval, which will shorten APD in the following beat, causing repeated short–long cycles related to the availability of depolarising channels.
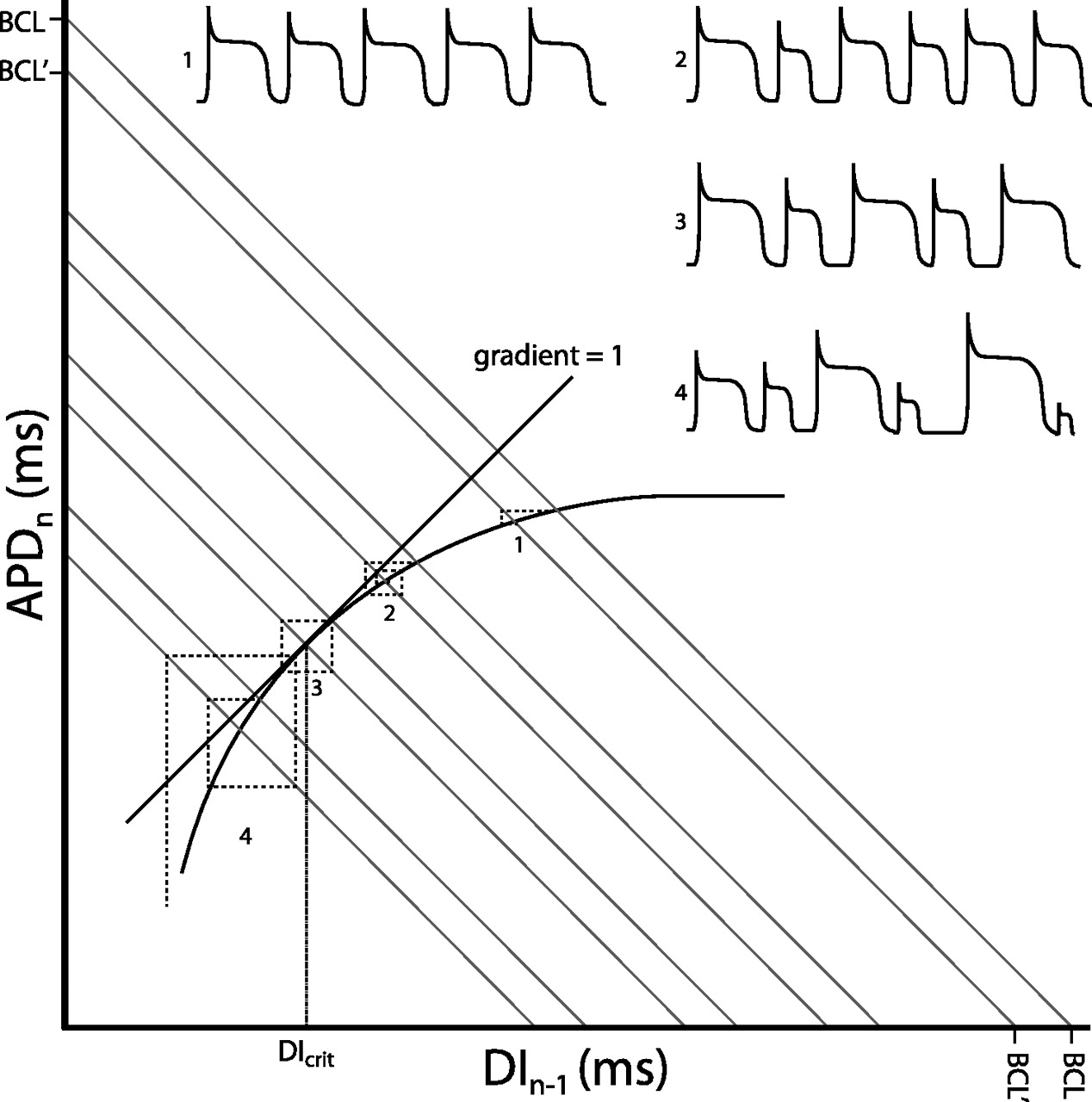
Alternans phenomena have classically been analysed by the construction of restitution curves relating APD to the preceding diastolic interval as heart rate is varied.17 The ‘restitution hypothesis’ states that transient APD alternans will occur when the slope of the restitution curve does not equal zero, becomes sustained when it is at unity and becomes unstable when the gradient exceeds one (figure 2). Relationships among restitution curve slopes, alternans and arrhythmogencity have been established in canine preparations made to model congenital LQTS type 218 and of genetic mouse models of LQTS type 3 and BrS.19 More recently, a predisposition of the RV for alternans has been found in the Scn5a+/− mouse model for BrS.20 Although concordant alternans itself may not be necessarily arrhythmogenic, it is a prerequisite for subsequent development of discordant alternans. This can have significant consequences on the spatial organisation of repolarisation across the ventricle, amplifying the heterogeneities of repolarisation present at baseline into pathophysiological heterogeneities of sufficient magnitude to produce conduction block and re-entrant excitation.

Restitution curve with action potential duration (APDn) plotted against the previous diastolic interval (DIn−1). Both would be in the order of milliseconds. The thick curve shows the typical monoexponential relationship this provides. Corresponding action potentials (APs) are shown for each numbered point. As the sum of the APD and DI gives the Basic Cycle Length, BCL, this gives the equation APD=−DI+BCL which is in the form of the linear relationship y=mx+c, with m the slope and c the vertical intercept of the function. Hence BCL represents the y-axis and x-axis intercept connected by a line with gradient of −1 (grey lines). Moving from a given BCL to another results in an iterative process which takes the APD to a new steady-state value. This is represented by the dashed lines. Each time a dashed line crosses the restitution function an AP is fired. At (1) the change in BCL results in a rapid iteration to a new steady-state value. As the gradient becomes steeper, the system takes longer to reach the steady-state value, with (2) showing a transient set of alternans. When the gradient of the restitution function reaches unity (3), the system enters a stable positive feedback, never reaching a steady-state value and producing sustained alternans. The DI at this point is known as the critical diastolic interval, DIcrit, and represents the break-point between stability and instability. Finally, surpassing the DIcrit (4) results in an unstable positive feedback, with AP durations spiralling away from each other. This is likely to result in the occurrence of discordant alternans and wavebreak, leading to arrhythmia.
Methods employed in basic electrophysiological studies of inherited cardiac arrhythmias
In common with many fields of medicine, investigation of inherited cardiac arrhythmias has relied on the translational cycle of identifying clinical problems via a genetic approach using familial studies, studying this genotype at the bench level in order to refine analysis techniques and to characterise the underlying mechanism, followed by a move back into the clinic in the form of risk stratification and drug trials. A significant contribution has been made from the utilisation of animal models including ferrets, rabbits, dogs, guinea pigs and rats, although they are all limited by the use of non-specific pharmacological agents to mimic the effects of genetic mutations. The introduction of genetically engineered mice has provided potentially more specific modelling opportunities. Despite differences in size, basal rate and relative contribution of repolarising K+ currents,21 they reliably replicate many of the human phenotypes. Structurally the sinoatrial node, atrioventricular node and atrioventricular bundles are similar in murine and human hearts and, importantly, the mouse shares similar depolarising Na+ currents to that of larger mammals including humans.22 Furthermore, both species have similar transmural differences in APD,23 share similar relationships between APD and refractory period24 and have near identical transmural conduction velocities.25 Genetic mouse models have yielded important findings concerning mechanisms underlying arrhythmogenicity in conditions including congenital LQT,26–28 BrS29 and CPVT.30
Channelopathies
Brugada syndrome
BrS is characterised electrocardiographically by right precordial ST elevation, negative T-waves and RV delay, and clinically by episodes of polymorphic VT and ventricular fibrillation (VF). While many genetic mutations have been associated with BrS, all result in a general imbalance of currents in favour of repolarisation over depolarisation. The most common and best studied of these is the loss-of-function mutation of the SCN5A gene, encoding the α-subunit of the voltage gated Na+ channel.
The cause of ST elevation in BrS and its strong linkage to VT/VF remains unresolved. Pathophysiological mechanisms responsible for ST segment changes must operate during the cardiac repolarisation phase, and these mechanisms must be based on INa reduction. The proposed mechanism which originally appeared to receive the widest support, both from experimental studies using a canine pharmacological ventricular preparation31 and some small clinical studies,32 ascribes BrS to a primary repolarisation disorder. The deep phase 1 notch in the epicardial AP, particularly prominent in the RV, renders it susceptible to the effects of a reduction in the Na+ current. The reduction in Na+ current establishes a steep voltage gradient across the RV wall due to short-circuiting of the epicardial AP resulting in extreme shortening. The imbalance of currents allows for reactivation of the RV epicardium by neighbouring regions of myocardium, with longer APs producing functional re-entry, referred to as phase 2 re-entry.
An alternative explanation for the ECG signature in BrS is based on conduction delay in the right ventricular outflow tract (RVOT).33 There is clinical evidence implicating conduction alterations, although this mainly employs non-invasive techniques such as echocardiography, signal-averaged ECG and body surface mapping.34 35 A single ex vivo study of the heart from a BrS patient has demonstrated local conduction delays in the RVOT, but in an absence of transmural differences in repolarisation.36 More recently, fractionated late potentials were found exclusively in the anterior aspect of the RVOT epicardium in BrS patients, and catheter ablation over this abnormal area resulted in normalisation of the ECG pattern and prevented VT/VF.37 So far, there is only one available clinical study that has showed both conduction and repolarisation disturbances.38
Recent studies using a heterozygotic Scn5a+/− mouse model have shed further light on possible pathological mechanisms of arrhythmogenesis. Affected mice demonstrate both depolarisation abnormalities, in the form of delayed conduction latencies29 and slowed activation patterns,39 and repolarisation abnormalities in the form of APD heterogeneities and steep restitution curves13 with an associated propensity to discordant alternans,20 which may act together to initiate arrhythmias (figure 3). Fibrosis and reduced connexin expression associated with ageing41 have also been demonstrated, which may contribute to the slowed conduction and furthermore may itself promote and maintain differences in repolarisation timing between the myocardial layers creating optimal conditions for re-entry. Thus, both structural changes and differences in ion channel kinetics together create optimal proarrhythmic conditions, and the disease can be regarded as a combination of a structural cardiomyopathy in conjunction with a functional channel anomaly.
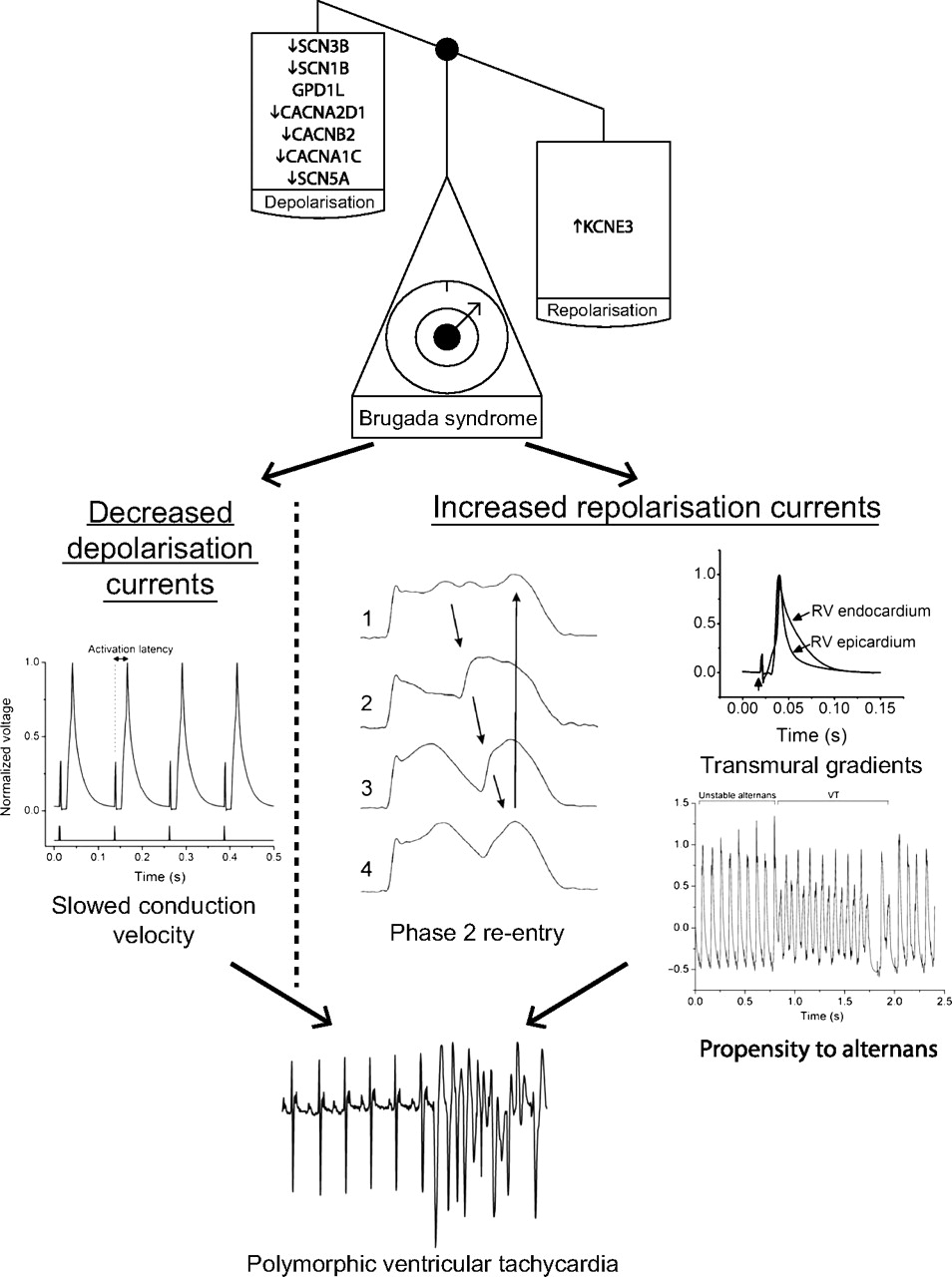
Mechanisms of arrhythmogenesis in Brugada syndrome from animal models. The scales demonstrate that its associated genetic mutations result in a loss of depolarising currents or a gain of repolarising currents. This in turn results in delayed conduction velocity, transmural action potential gradients, a greater propensity to alternans and potentially to phase 2 re-entry. The lowest trace shows an ECG with the initiation of a polymorphic ventricular tachycardia recapitulating the clinical phenotype. All traces shown are from mice except phase 2 re-entry which is from the canine wedge preparation. Adapted in part from Martin et al,29 Morita et al,40 Matthews et al20 and Martin et al,39 with permission.
Long QT syndrome
LQTS forms a genetically heterogeneous group of conditions, characterised by prolonged ventricular repolarisation and increased risk of ventricular arrhythmias, particularly torsade de pointes. In contrast to BrS, the mutations in LQTS result in a favouring of depolarising currents over repolarising ones. Evidence from several mouse models (for references see27) have implicated prolonged APD in two major problems generating an arrhythmic substrate (figure 4). First, increased refractory periods from channels that remain depolarised for greater periods create pockets of functional block, which may act as the focus for re-entry. Second, differences in dispersion of repolarisation across the myocardium may result in a functional re-entry acting as a trigger for torsade de pointes. EADs are also more common due to the extended time of depolarisation, which may in turn trigger polymorphic VT.

Mechanisms of arrhythmogenesis in long QT syndrome from animal models. The scales demonstrate that associated genetic mutations result in a gain of depolarising currents or a loss of repolarising currents. This in turn results in triggered activity in the form of early after-depolarisations, the formation of refractory pockets and the generation of abnormal transmural gradients. The lowest trace shows an ECG with the initiation of torsade de pointes recapitulating the clinical phenotype. All traces shown are from mice. Adapted in part from Thomas et al,42 Stokoe et al28 and Fabritz et al,43 with permission.
Catecholaminergic polymorphic ventricular tachycardia
CPVT presents as syncope or SCD due to the action of increased catecholamines during exercise or stress resulting in a bidirectional VT. The underlying mechanism of CPVT revolves around abnormal Ca2+ release from the SR, which is often due either to gain-of-function mutations in the gene encoding RyR2, a critical regulator of Ca2+ release from the SR for excitation–contraction coupling,44 or loss of function mutations in calsequestrin, a SR calcium buffering protein.45
Such abnormal Ca2+ release may cause DADs, similar to digitalis toxicity where baseline Ca2+ is also elevated, resulting in an arrhythmic trigger46 (figure 5). Data from in vitro studies show that CPVT-associated RyR2 and CASQ2 mutations8 49 cause cytosolic Ca2+ overload which generates a net inward current. Several transgenic mouse models of CPVT have been developed, harbouring mutations in either RyR230 or in CASQ2,50 which demonstrate abnormal Ca2+ transients and bidirectional VT. These studies have provided evidence that the transient inward current may underlie the DADs that trigger abnormal beats in this condition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanisms of arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia from animal models. The genetic mutations result in an increase in cytosolic Ca2+ which in turn causes the scales to tip in favour of depolarising currents due to the electrogenic nature of the Na+/Ca2+ exchanger. If sufficient, this may lead to triggered activity in the form of delayed after-depolarisations. The lowest trace shows an ECG with the initiation of a bidirectional ventricular tachycardia recapitulating the clinical phenotype. All traces shown are from mice. Adapted in part from Cerrone et al47 and Cerrone et al,48 with permission.
Other syndromes
While the majority of cases of SCD in the absence of structural heart abnormalities are caused by the three syndromes mentioned above, there are a number of other related syndromes that have come to light. Short QT syndrome, phenotypically characterised by short APDs and hence QT intervals, can be caused by gain-of-function mutations in K+ channels51 or loss of function mutations in Ca2+ channels.52 Studies in a canine pharmacological wedge preparation have implicated increased transmural repolarisation gradients in the arrhythmogenic mechanism.53
Early repolarisation (ER) disease is characterised by ventricular arrhythmias and sudden death in the presence of an ER pattern on the ECG as characterised by J-point elevation and prominent T waves.54 While preliminary evidence from canine studies suggests that increased transmural differences in early phases of the AP may be responsible,55 the exact mechanism for ER is still unknown. In rare cases, mutations in genes causing either a decrease in inward Ca2+ currents56 or an increase in outward K+ currents57 have been identified; however, mutations have not been identified in the majority of cases. Given the high frequency of the genetic background underlying the ER pattern in the population, the disease is probably polygenic and influenced by environmental factors.
Progressive cardiac conduction disease (PCCD) encompasses another group of conditions that, while most commonly manifesting as bradycardia, may also cause SCD. The first gene to be associated with PCCD was SCN5A, and indeed some SCN5A mutations may be associated with more complex phenotypes combining characteristics of BrS, PCCD and LQT3, the so-called ‘overlap syndromes’.58 More recently, PCCD has been associated with altered expression of genes encoding other proteins involved in impulse propagation, including those responsible for calcium-activated ion channels and cytoskeletal components. Mouse studies have indicated that the resulting phenotype appears to develop from a combination of these genetic factors, environmental modifiers and other physiologic determinants, including ageing.41
Conclusion
A clearer understanding of the underlying causes of inherited arrhythmias has arisen as a result of basic research, particularly from engineered mice modelling genetic abnormalities. In particular, as demonstrated in all of the inherited disorders, imbalances of current in favour of either depolarisation, such as in the LQTS and CPVT, or repolarisation, such as in BrS and short QT syndrome, in a spatially or temporally heterogeneous fashion may result in foci for re-entrant circuits, potentially giving rise to major arrhythmia. Linking inherited arrhythmogenic syndromes to their genetic and molecular basis offers the practitioner tools for accurately diagnosing rare disorders and provides novel markers for assessing risk of SCD. Recognising the fundamental defects in channelopathies may also provide the basis for new strategies of treatment, including tailored pharmacotherapy and gene therapy.
References
Footnotes
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.