Article Text

Abstract
Background The optimal duration of clopidogrel treatment, particularly following drug-eluting stent (DES) implantation, remains contentious. Previous studies have observed a clustering of adverse events following clopidogrel cessation 1 year after DES, the aetiology of which is poorly understood.
Objective To investigate, in the prospective CESSATION study, the effect of clopidogrel withdrawal at 1 year after DES implantation on (i) arachidonic acid (AA)- and adenosine diphosphate (ADP)-induced platelet aggregation, and (ii) biomarkers of vascular inflammation, including soluble CD40 ligand (sCD40L), high-sensitivity C-reactive protein (hsCRP) and interleukin 6 (IL-6).
Methods and results The prospective CESSATION study was undertaken in 33 patients receiving aspirin and due to discontinue clopidogrel 1 year after DES. Platetet reactivity was measured using short thromboelastography, and compliance with aspirin determined from serum thromboxane B2 (TXB2) levels. Venesection was performed at 4 weeks and 24 h before, and at 24 h, 48 h, 1, 2 and 4 weeks after, clopidogrel cessation. Following clopidogrel withdrawal, there was (i) a predictable increase in ADP-induced platelet aggregation (ii) an unexpected significant increase in AA-induced platelet aggregation (iii) a decline in IL-6 and hsCRP at 1 week and 4 weeks respectively; and (iv) a non-significant increase in sCD40L at 4 weeks TXB2 levels were consistently suppressed, indicating complete inhibition of cyclo-oxygenase-1 by aspirin.
Conclusion An aspirin-independent, time-dependent increase in AA-induced platelet activation following clopidogrel withdrawal in patients with a DES has been described. New insights into a potential mechanism for the observed clustering of adverse events that occur early after clopidogrel cessation have been provided. These findings raise the question as to whether AA-induced clotting is an appropriate test of aspirin sensitivity.
- Aspirin
- clopidogrel cessation
- drug eluting stent
- inflammation
- platelet aggregation
- coronary artery disease
- interventional cardiology
- platelets
- platelet activation
- NSAIDS
- endothelium
- endothelin
- antiplatelet treatment
- coronary stenting
- coronary angioplasty (PCI)
- stable angina
- NSTEMI
- angina treatment
- angina—unstable
- acute coronary syndrome
- cardiac ultrasound
- stents
- echocardiography
- EBM
- angioplasty offsite
- angiography
- platelet and angioplasty
- coronary syndromes
- acute ischaemic syndromes
- acute myocardial infarction
- angina
Statistics from Altmetric.com
- Aspirin
- clopidogrel cessation
- drug eluting stent
- inflammation
- platelet aggregation
- coronary artery disease
- interventional cardiology
- platelets
- platelet activation
- NSAIDS
- endothelium
- endothelin
- antiplatelet treatment
- coronary stenting
- coronary angioplasty (PCI)
- stable angina
- NSTEMI
- angina treatment
- angina—unstable
- acute coronary syndrome
- cardiac ultrasound
- stents
- echocardiography
- EBM
- angioplasty offsite
- angiography
- platelet and angioplasty
- coronary syndromes
- acute ischaemic syndromes
- acute myocardial infarction
- angina
Introduction
Dual antiplatelet treatment with aspirin and clopidogrel is recommended in all patients undergoing percutaneous coronary intervention (PCI) and following acute coronary syndromes (ACS) to reduce the risk of ischaemic events and cardiovascular mortality. Current clinical guidelines recommend lifelong aspirin and clopidogrel for up to 1 year, after which time clopidogrel is abruptly withdrawn.1 The optimal duration of clopidogrel treatment, particularly after drug-eluting stent (DES) implantation, remains a contentious issue and is the focus of continuing clinical studies.2–4 Ho et al5 6 reported a clustering of adverse clinical events within the first 90 days of clopidogrel withdrawal in patients with ACS treated either medically or with PCI. It has been suggested that this may be due to a ‘rebound’ prothrombotic and/or proinflammatory response that occurs following cessation of chronic clopidogrel treatment.7 However, the causative mechanism behind this observed response and therefore a potential strategy to attenuate this effect has not been fully elucidated.
Previous clinical studies have investigated the effect of clopidogrel withdrawal on (i) inflammatory biomarkers alone8; (ii) platelet reactivity alone9 and (iii) inflammatory biomarkers and platelet reactivity in a diabetic population alone.10 The results were conflicting and warrant further investigation. Furthermore, previous data also suggest that there is an interaction between clopidogrel and the arachidonic acid (AA)-induced pathway of platelet reactivity.11–13 Such a mechanism might be of considerable importance at a time when clopidogrel is abruptly withdrawn, because it may enhance the inevitable increase in adenosine diphosphate (ADP)-mediated platelet reactivity.
Thus, the effect of clopidogrel cessation on both biomarkers of inflammation and platelet reactivity at multiple time-points is unknown. Importantly, the effect of clopidogrel cessation on AA-induced pathways of platelet reactivity, currently thought to be predominantly influenced by aspirin, is unknown. Therefore, the CESSATION study investigated the effect of clopidogrel withdrawal at 1 year after DES implantation on (i) AA- and ADP-induced platelet aggregation, and (ii) biomarkers of vascular inflammation, including soluble CD40 ligand (sCD40L), high-sensitivity C-reactive protein (hsCRP) and interleukin 6 (IL-6).
Methods
Study population
The study was approved by the Southampton and South West Hampshire Research Ethics Committee and conducted in accordance with the Declaration of Helsinki. Study participants were identified by review of a database from a single regional cardiothoracic centre. Between February and June 2010, 38 patients receiving low-dose aspirin treatment who had undergone PCI with DES 11 months earlier and were due to stop clopidogrel at 1 year were prospectively enrolled after obtaining written informed consent. Subjects were excluded if they were taking non-steroidal anti-inflammatory drugs, steroids or anticoagulant treatment.
Study design
Blood samples were taken at the following prespecified time-points during the study period: (i) 4 weeks and 24 h before clopidogrel cessation, and (ii) 24, 48 h, 1, 2 and 4 weeks after clopidogrel cessation. At each time-point, platelet reactivity was measured using short thromboelastography (TEG)11 14–19 and thromboxane B2 (TXB2), IL-6, hsCRP and sCD40L serum levels were measured.
Blood sampling and analysis
Venesection was performed from the antecubital fossa, and the first 2 ml discarded. Blood was then drawn into a 6 ml lithium heparin Vacutainer for TEG, a 5 ml serum-SST Vacutainer for TXB2 analysis and a 4 ml EDTA Vacutainer for IL-6, CD40L and hsCRP analysis. Serum-SST and EDTA tubes were centrifuged at 3500 rpm for 10 min and immediately frozen at −80°C for future batch analysis.
Thromboelastography
Platelet reactivity was measured using the modified TEG platelet mapping system (Haemonetics Corp, Massachusetts, USA), according to the manufacturer's instructions. Modified TEG uses four channels to detect the effects of antiplatelet treatment acting via the AA and ADP pathways. The four channels contain (1) kaolin, (2) activator F (a mixture of reptilase and factor XIIIa), (3) activator F and AA (1 mm), (4) activator F and ADP (2 μm). Whole blood (360 μl) is pipetted into each channel, which consists of an oscillating cup into which a pin is suspended. As the blood clots, changes in its viscoelasticity are transmitted to the pin and the resulting torque generates an electrical signal producing a TEG trace which is a graphic representation of the speed and strength of clot formation.20 The following parameters are derived from the TEG trace:
Maximum amplitude (MA): the MA is derived directly from the automated TEG analytical software and is representative of maximal clot strength.
Area under the response curve at 15 min (AUC15): AUC15 incorporates both the rate of rise (speed of clot formation) and the maximum amplitude (strength of the clot) of the TEG trace. It is calculated using a specially developed software program, National Instrument Labview 7.0 (Areafinder 2:1). AUC15 has been shown to correlate well with MA in assessment of responses to antiplatelet treatment.14 The AUC15 TEG method, known as short TEG, has been well validated by this group and is described in detail elsewhere.11 14–19 Recent data from this group19 have shown that the novel parameter AUC15 is not only easily reproducible with minimal intra- and interindividual variability, but also correlates well with VerifyNow in the assessment of responses to aspirin and clopidogrel (AUC15 AA channel and VerifyNow aspirin response units, correlation coefficient r=0.701; p<0.001 and AUC15 ADP channel and VerifyNow P2Y12 reactivity units, correlation coefficient r=0.609, p<0.001).
Serum thromboxane B2
In order to assess treatment compliance and activity of aspirin, serum TXB2 levels were determined using a selective, competitive ELISA (Cayman Chemicals, Ann Arbor, Michigan, USA), according to the manufacturer's instructions. Samples were diluted 1:10 in diluent and assayed parallel to known TXB2 standards and a maximum binding control. The percentage binding of known standards was calculated in reference to the maximum binding (zero TXB2) control wells, plotted against the logarithm of concentration and analysed by non-linear regression using a four-parameter logistical fit model. Unknown samples were expressed similarly, interpolated from this standard curve and corrected for dilution.
Inflammatory markers
sCD40L, hsCRP and IL-6 were measured in duplicate using commercially available ELISA kits from R&D Systems (Abingdon, UK), according to the manufacturer's instructions.
Statistical analysis
This is an exploratory study. There are no data available on the effect of clopidogrel cessation on all three parameters (ie, platelet reactivity, TXB2 and inflammatory biomarkers) in this study population. Data are presented as the mean change from baseline with 95% CIs, unless otherwise stated. The baseline time-point refers to 24 h before clopidogrel cessation. Significance between time-points was determined using paired t tests with a p value of <0.05 considered to represent statistical significance. Owing to the exploratory nature of the study no adjustments for multiple testing were made. Statistical analysis was performed using SPSS version 17.0 software, Microsoft Excel and Prism v5.0 (GraphPad software, La Jolla, California, USA).
Results
A total of 38 patients were enrolled in the study. Four patients withdrew pre-maturely and one patient was excluded because he chose not to discontinue clopidogrel as required. Thus, study data are presented on 33 patients. Baseline characteristics and demographics of the study population are provided in table 1. The participants were predominantly male (82%), with a mean age (SD) of 65.9 (8.2) years. Cardiac risk factors included hyperlipidaemia (58%), family history of ischaemic heart disease (52%), hypertension (30%), active smoking (12%) and diabetes (3%). All participants were receiving aspirin 75 mg, clopidogrel 75 mg and statin treatment. Eight (24%) patients were receiving proton pump inhibitors and six (18%) were receiving calcium channel blockers. Mean total duration (SD) of clopidogrel treatment after PCI was 373 (7.1) days.
Baseline demographics (n=33)
Clinical events
One patient was admitted with stent thrombosis 8 days after clopidogrel cessation. Clopidogrel was therefore recommenced in accordance with ACS treatment guidelines. Subsequent coronary angiography confirmed an occluded right coronary artery which was successfully treated with PCI. We have included study data up until the fifth time-point in this patient (ie, just before his presentation with stent thrombosis).
Thromboxane B2 levels
TXB2 was suppressed consistently at all time-points, confirming effective inhibition of cyclo-oxygenase (COX)-1 by aspirin throughout the study. Mean (SD) serum TXB2 concentrations were 122.7±203.8 pg/ml at 4 weeks before, 137.7±317.9 pg/ml at 24 h before, 94.3±74.4 pg/ml at 24 h after, 61.8±47.6 pg/ml at 48 h after, 129.7±125.4 pg/ml at 1 week after, 164.3±232.7 pg/ml at 2 weeks after and 103.5±85.4 pg/ml at 4 weeks after clopidogrel cessation. The change in mean serum TXB2 from baseline was −43.4 (95% CI −156.2 to 69.4, p=0.439) at 24 h after, −75.9 (95% CI −187.5 to 35.7, p=0.176) at 48 h after, −8.1 (95% CI −103.3 to 87.2, p=0.864) at 1 week after, 26.5 (95% CI −111 to 164.1, p=0.697) at 2 weeks after and −36.5 (95% CI −155.8 to 82.8, p=0.537) at 4 weeks after clopidogrel cessation.
ADP-induced platelet aggregation
Following clopidogrel withdrawal, there was a significant and time-dependent increase in ADP-induced platelet aggregation measured using s-TEG AUC15 (figure 1). The change in mean AUC15 from baseline was 128.3 (95% CI 71.9 to 184.7, p<0.001) at 24 h after, 193.2 (95% CI 134 to 252.4, p<0.001) at 48 h after, 271.3 (95% CI 194 to 348.6, p<0.001) at 1 week after, 295.8 (95% CI 212.7 to 378.9, p<0.001) at 2 weeks after and 280.4 (95% CI 203.4 to 357.4, p<0.001) at 4 weeks after clopidogrel cessation (table 2). The percentage change in mean AUC15 from baseline was 15.6% at 24 h after clopidogrel cessation and increased to 33% at 1 week after clopidogrel cessation.

Area under the curve at 15 min (AUC15) of the adenosine diphosphate (ADP) channel. Error bars represent mean with 95% CIs. The p value given (***p<0.001) is for comparison with the baseline time-point at 24 h before clopidogrel cessation. Although the x-axis time-points are equally spaced on the graph, the actual time intervals between any two points vary.
Change in ADP- and AA-induced platelet aggregation following clopidogrel cessation measured using s-TEG area under the curve at 15 min (AUC15)
AA-induced platelet aggregation
Following clopidogrel withdrawal, there was a significant increase in AA-induced platelet aggregation measured using s-TEG AUC15 (figure 2). The change in mean AUC15 from baseline was 129 (95% CI 46.2 to 211.8, p=0.005) at 24 h after, 171.6 (95% CI 74.9 to 268.3, p=0.002) at 48 h after, 242.9 (95% CI 146.7 to 339.1, p<0.001) at 1 week after, 316.2 (95% CI 200.7 to 431.7, p<0.001) at 2 weeks after and 235.7 (95% CI 115.3 to 356.1, p=0.001) at 4 weeks after clopidogrel cessation (table 2). The percentage change in mean AUC15 from baseline was 31.5% at 24 h after clopidogrel cessation and increased to 59.4% at 1 week after clopidogrel cessation.

Area under the curve at 15 min (AUC15) of the arachidonic acid (AA) channel. Error bars represent mean with 95% CIs. The p values given (**p<001; ***p<0.001) are for comparison with the baseline time-point at 24 h before clopidogrel cessation. Although the x-axis time-points are equally spaced on the graph, the actual time intervals between any two points vary.
Concomitant medical treatment and platelet reactivity
Patients receiving regular proton pump inhibitors (n=8) and calcium channel blockers (n=6) did not show a trend towards higher residual AA- or ADP-induced platelet reactivity before clopidogrel cessation. Furthermore, the increase in platelet aggregation after clopidogrel withdrawal did not amplify in this group.
Inflammatory biomarkers
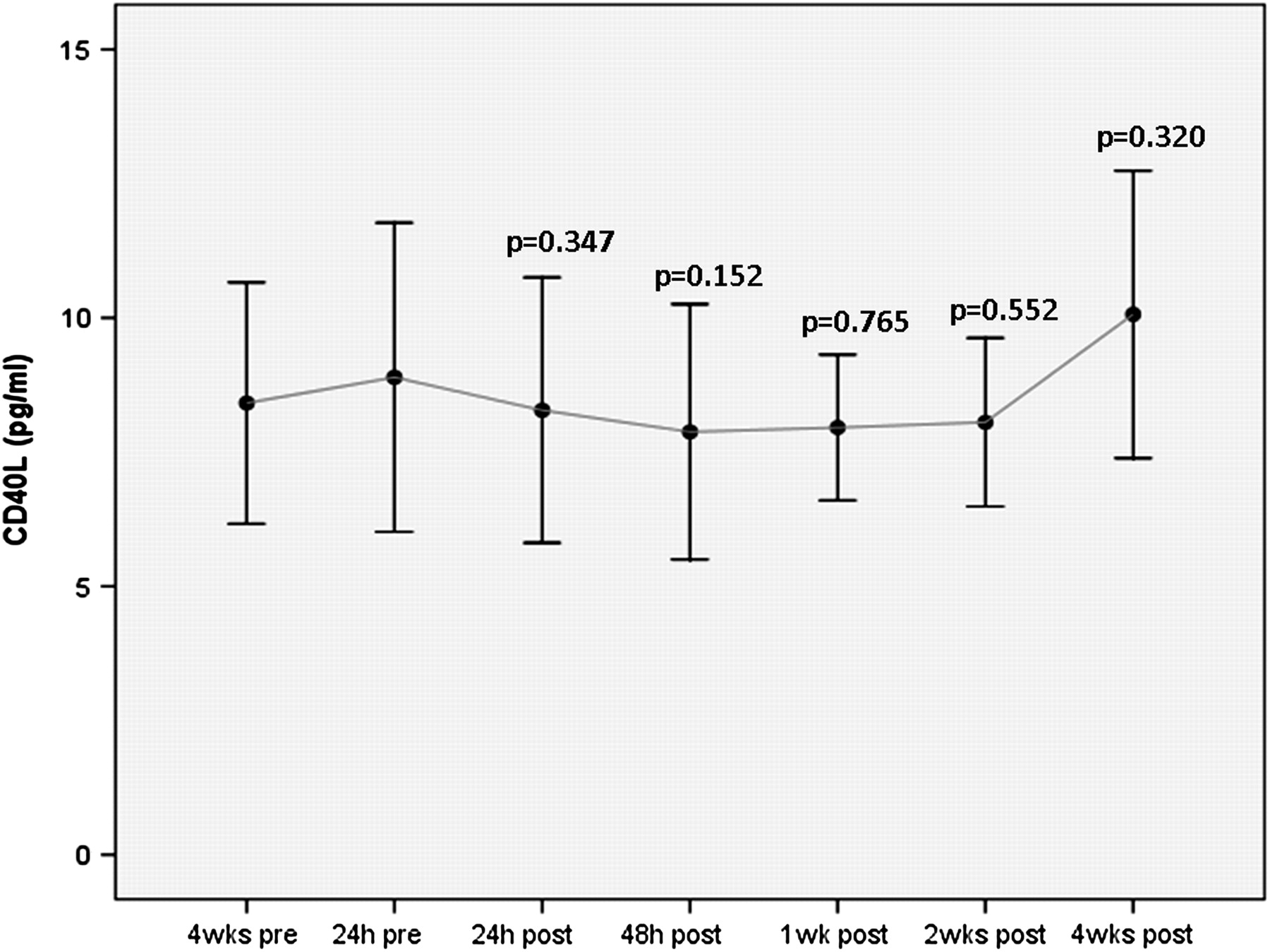
There was a decline in mean IL-6 at 1 week after clopidogrel cessation (p=0.006), a decline in hsCRP at 4 weeks after clopidogrel cessation (p= 0.033) and an insignificant increase in sCD40L at 4 weeks after clopidogrel cessation (p=0.32) (figures 3–5, table 3).
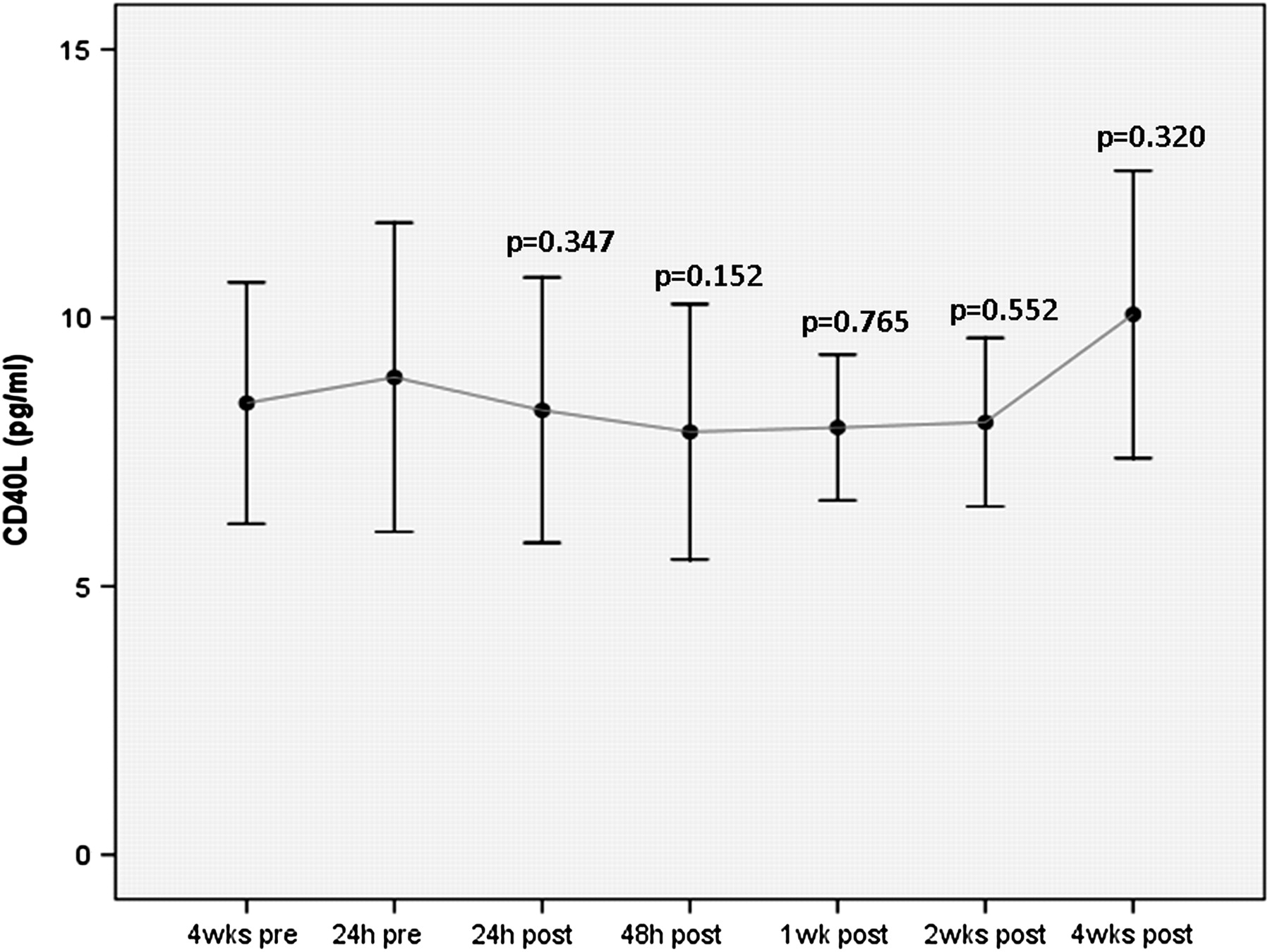
Soluble CD40 ligand (sCD40L) before and after clopidogrel cessation. Error bars represent mean with 95% CIs. The p values given are for comparison with the baseline time-point at 24 h before clopidogrel cessation. Although the x-axis time-points are equally spaced on the graph, the actual time intervals between any two points vary.
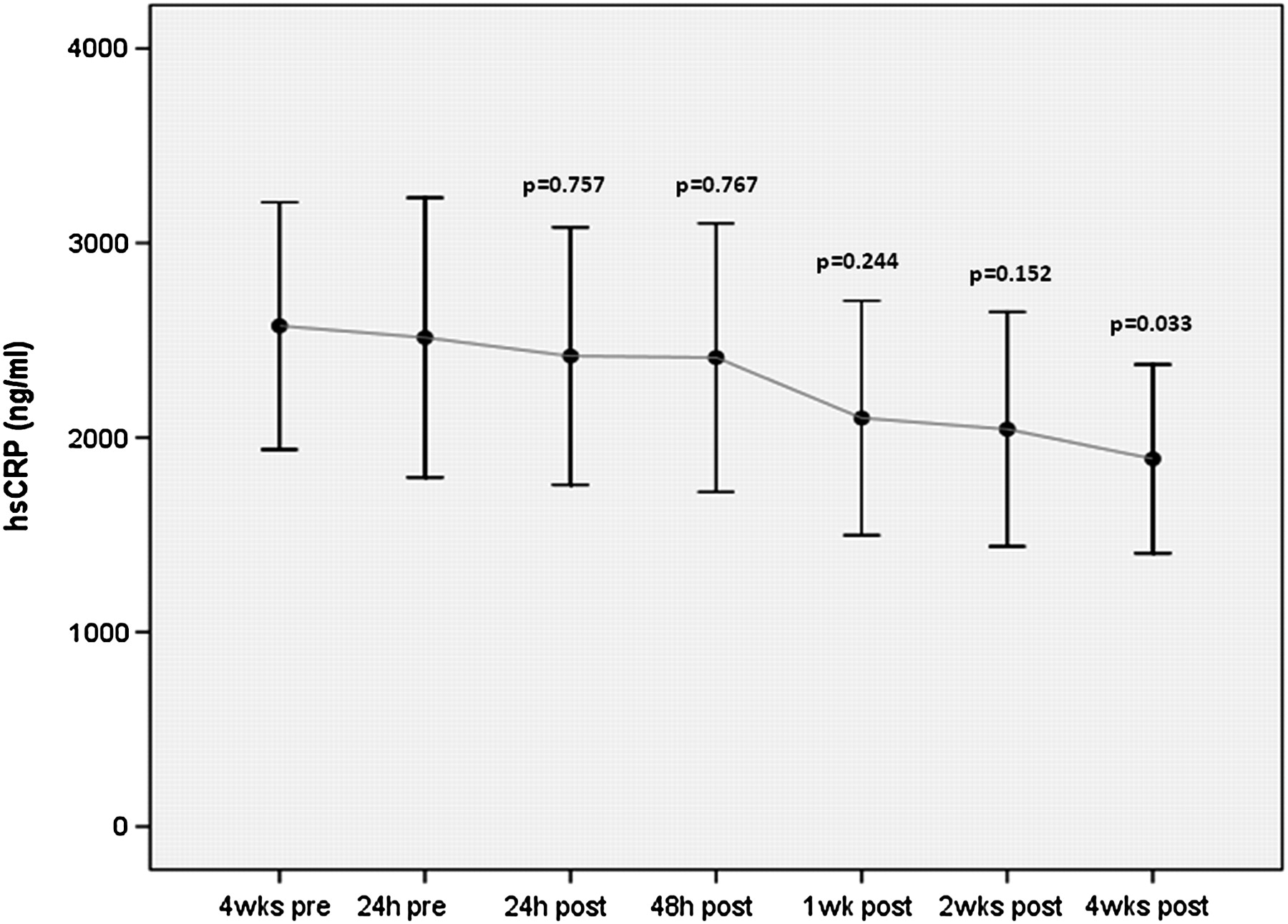
High-sensitivity C-reactive protein (hsCRP) before and after clopidogrel cessation. Error bars represent mean with 95% CIs. The p values given are for comparison with the baseline time-point at 24 h before clopidogrel cessation. Although the x-axis time-points are equally spaced on the graph, the actual time intervals between any two points vary.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Interleukin-6 before and after clopidogrel cessation. Error bars represent mean with 95% CIs. The p values given are for comparison with the baseline time-point at 24 h before clopidogrel cessation. Although the x-axis time-points are equally spaced on the graph, the actual time intervals between any two points vary. *At this time point there was a solitary inexplicably high IL-6 value observed in one patient.This was approximately 100-fold greater than all other IL-6 measurements. As described in the results section of the text, this is likely to be a spurious result and has therefore been excluded from the data analysis.
Change in inflammatory markers soluble CD40 ligand (sCD40L), interleukin 6 (IL-6) and high-sensitivity CRP (hsCRP) following clopidogrel cessation
Of note, there was a solitary inexplicably high IL-6 value at 2 weeks after clopidogrel cessation in one patient which was approximately 100-fold greater than all other IL-6 measurements. There was no accompanying significant rise in any of the other inflammatory markers at this time-point and, furthermore, the patient did not report any illness or adverse event at any time during the study period. Thus, this solitary IL-6 measurement is likely to be spurious and has been excluded from the analysis.
Discussion
This study has the novel finding that, when clopidogrel is withdrawn 1 year after DES implantation, there is a significant aspirin-independent, time-dependent increase in AA-induced platelet reactivity in addition to a predictable time-dependent increase in ADP-induced platelet reactivity as measured by short TEG. Further, clopidogrel withdrawal is associated with a decline in IL-6 and hsCRP, results which are unexpected given its anti-inflammatory reputation.
These findings provide new insights into a potential mechanism for the observation that adverse events cluster early after clopidogrel withdrawal. Also, our results further support accumulating evidence that clopidogrel influences AA pathways and thereby potentiates the effect of aspirin.11–13 A secondary but important issue that arises is whether AA-induced platelet aggregation is an appropriate test for aspirin sensitivity. It is well established that aspirin achieves its antithrombotic effects through inactivation of COX-1 and thus prevents generation of TXA2 and its stable metabolite TXB2. Serum TXB2 analysis is the most specific pharmacological test to evaluate the effect of aspirin on platelets and, furthermore, is a marker of compliance with aspirin treatment. In this study, despite a significant increase in AA-induced platelet reactivity after clopidogrel cessation, TXB2 levels were consistently suppressed, indicating complete inhibition of platelet COX-1 by aspirin. This observation therefore excludes aspirin treatment failure as an explanation for the increase in AA-induced clotting and suggests that there is an alternative clopidogrel-mediated mechanism responsible for the observed AA pathway response. Similar observations were reported by Frelinger et al21 who demonstrated a P2Y12-dependent but COX-independent pathway of residual AA-induced platelet activation in a cohort of 700 consecutive aspirin-treated patients undergoing PCI. These findings demand further investigation by large-scale clinical studies to elicit the precise mechanisms by which clopidogrel achieves its antithrombotic effect. Specifically, the extent to which an effective P2Y12 inhibitor might achieve blockade of the AA–TXA2 pathway requires further assessment. Conceivably, if such effects were sufficiently potent, the addition of aspirin would be obsolete. This remains speculative, pending further data.22
At the very least, our data suggest that measurement of AA-induced platelet aggregation may not be a reliable test for measuring clinical response to aspirin, given that this parameter changed in the patients in this study in whom there was complete suppression of TXB2 levels. This is potentially an explanation for the apparently high levels of functional ‘aspirin resistance’ reported in studies using AA-mediated tests. For example, a study investigating the incidence of ‘aspirin resistance’ in 45 patients with stroke reported a rate of 67% measured using the AA-specific unmodified TEG method.23 Serum TXB2 levels were not measured in that study.
Elevated inflammatory biomarkers have been linked to poor outcome after PCI and increased risk of adverse cardiovascular events in patients with ACS.24–26 Although the anti-inflammatory properties of clopidogrel in acute treatment of disease have been well described,27–30 data about its anti-inflammatory effects in patients with stable coronary artery disease receiving chronic treatment are inconsistent and conflicting. For example, one randomised controlled study showed that long-term clopidogrel in patients with stable coronary artery disease significantly inhibited the production of sCD40L but had no effect on hsCRP levels.31 Similar apparently discordant findings in a different patient group were also observed in the ELAPSE study,32 which showed a significant decline in sCD40L one year after PCI in patients receiving chronic clopidogrel, as well as an increase in soluble P selectin and serum IL-18 levels and no change in hsCRP compared with baseline.
In this CESSATION study, clopidogrel withdrawal 1 year after DES resulted in a decline in mean IL-6 at 1 week after (p=0.006), a decline in hsCRP at 4 weeks after (p= 0.033) but a non-significant increase in sCD40L at 4 weeks after clopidogrel cessation (p=0.32). Our findings are consistent with observations from the DECADES study,8 which also investigated the effect of clopidogrel cessation on biomarkers of inflammation 1 year after DES but did not examine the effect on platelet reactivity. That study reported a significant increase in sCD40L levels 4 weeks after clopidogrel withdrawal (p<0.001) but an unexplained and apparently inconsistent decrease in hsCRP levels 1 week after cessation of clopidogrel (p=0.008). Although the sCD40L findings from this and the DECADES study suggest that clopidogrel may have important anti-inflammatory properties, new questions are generated by our data: does chronic clopidogrel treatment upregulate certain inflammatory biomarkers and is the observed declining trend in hsCRP and IL-6 directly related to loss of the platelet inhibitory effect on the ADP and AA pathways associated with clopidogrel cessation?
Study limitations
Our study is limited by its small sample size. In addition, owing to the design of study we were unable to measure baseline pretreatment levels of platelet reactivity and inflammatory biomarkers. Furthermore, although it might have been useful to measure platelet reactivity using more than one laboratory assay of platelet function, previous studies from this group and others have shown strong correlation between TEG AUC15 and VerifyNow18 19 and between TEG and the historical ‘gold standard’ method optical aggregometry33 34 for measurement of response to antiplatelet treatment.
Conclusion
Our study demonstrates a significant time-dependent increase in ADP- and AA-induced platelet reactivity and a decline in IL-6 and hsCRP following clopidogrel withdrawal at 1 year in patients with a DES. This raises important clinical questions that demand further investigation. First, AA-induced clotting may not be the most appropriate test to measure the antiplatelet effect of aspirin, which means the relatively high rates of aspirin ‘resistance’ reported in previous clinical studies should be interpreted with caution and, at the very least, should include serum TXB2 measurements in parallel. Second, the time-dependent increase in AA-induced clotting observed in this study following clopidogrel withdrawal lends support to the accumulating evidence which suggests that clopidogrel exerts some of its antiplatelet effects via the AA pathway in an aspirin-independent fashion. However, it remains uncertain to what extent clopidogrel potentiates aspirin and whether the antithrombotic effect of aspirin is rendered partially or even completely redundant in the presence of clopidogrel or other more potent P2Y12 agents. This study raises the question as to whether clopidogrel should be withdrawn abruptly 1 year after DES or whether tapered withdrawal or discontinuation of aspirin instead may offer greater clinical benefit. Large-scale clinical outcome studies are clearly needed to answer these clinically relevant questions.
Acknowledgments
The authors would like to thank Wessex Heartbeat, a registered charity based at Southampton General Hospital that supports the work of the Wessex Cardiac Centre, for providing funding for the inflammatory biomarker analysis of this study.
References
Footnotes
Funding This work was supported by unrestricted grants from Haemonetics and Medtronic UK.
Competing interests NC has received unrestricted research funding from Haemonetics, Medtronic, Boston Scientific and Pfizer. He has also received speaker/consulting fees from Boston Scientific, Abbott, Medtronic, AstraZeneca, Eli Lilly and Cordis. TW has received unrestricted research funding and speaker/consulting fees from AstraZeneca. JM has received honoraria and consulting fees from Medtronic and St. Jude. The remaining authors report no conflicts of interest.
Ethics approval The Southampton and South West Hampshire Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.