Article Text

Abstract
Objective The inflammatory cytokine, tumour necrosis factor α (TNF-α), exerts deleterious cardiovascular effects. We wished to determine the effects of TNF-α antagonism on endothelial function and platelet activation in patients with acute myocardial infarction.
Design and setting and patients A double-blind, parallel group, randomised controlled trial performed in a tertiary referral cardiac centre. 26 patients presenting with acute myocardial infarction randomised to receive an intravenous infusion of etanercept (10 mg) or saline placebo.
Main outcome measures Leucocyte count, plasma cytokine concentrations, flow cytometric measures of platelet activation and peripheral vasomotor and fibrinolytic function were determined before and 24 h after study intervention.
Results Consistent with effective conjugation of circulating TNF-α, plasma TNF-α concentrations increased in all patients following etanercept (254±15 vs 0.12±0.02 pg/ml; p<0.0001), but not saline infusion. Etanercept treatment reduced neutrophil (7.4±0.6 vs 8.8±0.6×109 cells/l; p=0.03) and plasma interleukin-6 concentrations (5.8±2.0 vs 10.6±4.0 pg/ml; p=0.012) at 24 h but increased platelet–monocyte aggregation (30±5 vs 20±3%; p=0.02). Vasodilatation in response to substance P, acetylcholine and sodium nitroprusside, and acute tissue plasminogen activator release were unaffected by either treatment (p>0.1 for all).
Conclusions Following acute myocardial infarction, etanercept reduces systemic inflammation but increases platelet activation without affecting peripheral vasomotor or fibrinolytic function. We conclude that TNF-α antagonism is unlikely to be a beneficial therapeutic strategy in patients with acute myocardial infarction.
- MYOCARDIAL ISCHAEMIA AND INFARCTION (IHD)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The relationship between inflammatory disorders and atherosclerosis has been recognised for over a century, and there now exists clear evidence to support the concept that immune responses are critical in the initiation, progression and destabilisation of atherosclerosis. T lymphocytes and monocytes are present within stable and unstable atherosclerotic plaques, and act to mediate both atherogenesis and plaque destabilisation.1 The inflammatory nature of atherosclerosis is exemplified by the high incidence of cardiovascular disease in patients with chronic inflammatory conditions, such as rheumatoid arthritis2 and systemic lupus erythematosis,3 independent of traditional cardiovascular risk factors. Similarly, acute inflammation, such as that occurring in the context of infections of the respiratory and urinary tract, is temporally associated with an increased incidence of adverse cardiovascular events such as myocardial infarction and stroke.4 Elevated concentrations of inflammatory mediators predict the occurrence of adverse cardiovascular events, even in apparently healthy low risk individuals without overt cardiovascular disease.5 Despite the longstanding recognition of the importance of inflammation in the development of atherosclerosis and the precipitation of thrombotic events, anti-inflammatory therapy for the treatment of atherosclerosis has received little attention. However, targeted anti-inflammatory therapy is a highly attractive concept, particularly for the treatment of acute vascular inflammation, as this is a universal component of the development of acute coronary syndromes.
Tumour necrosis factor α (TNF-α) is a pro-inflammatory cytokine secreted by activated macrophages and T cells, and is instrumental in the mediation of vascular inflammation. TNF-α receptors are expressed on all critical cellular effectors of atherogenesis, namely, vascular endothelial and smooth muscle cells, and activated leucocytes such as T lymphocytes and macrophages.6 TNF-α production is deleterious to vascular homeostasis through the induction of endothelial surface adhesion molecule expression mediating platelet–leucocyte interactions, cellular aggregation and thrombus formation.7 TNF-α production is increased in unstable atherosclerotic plaques8 and is detectable in increased concentrations in patients with acute myocardial infarction (AMI).9 Furthermore, plasma TNF-α concentrations are predictive of death and recurrent ischaemic events following AMI.10 We have previously demonstrated that TNF-α mediates vascular inflammation with potentially beneficial pro-fibrinolytic effects; however, it is associated with adverse inhibitory effects on endothelium-dependent vasodilatation.11 Consistent with these deleterious effects, antagonism of the TNF-α receptor has been demonstrated to improve endothelium-dependent vasodilatation in a variety of clinical settings such as congestive cardiac failure,12 acute systemic vasculitis13 and rheumatoid arthritis.14 The positive vascular effects of TNF-α antagonism may therefore be beneficial in the management of acute coronary syndromes, which are strongly associated with endothelial dysfunction. We therefore studied the effects of the TNF-α receptor antagonist, etanercept, on peripheral vasomotor and fibrinolytic function, and platelet activation in patients presenting with recent AMI.
Methods
Subjects
The study was performed with the approval of the local research ethics committee in accordance with the Declaration of Helsinki and with the written informed consent of all participants. Inclusion criteria were a typical history of myocardial ischaemia lasting more than 20 min within 24 h of hospitalisation with ischaemic electrocardiographic changes and an elevated troponin-I concentration (>0.2 µg/l). Patients were excluded in the event of significant comorbidity, including active systemic inflammatory disorders, insulin-dependent diabetes mellitus and the use of anti-inflammatory drugs other than aspirin. Given the immunosuppressive effects of etanercept, exclusion criteria also included any history of recent or recurrent infection, tuberculosis or any opportunistic infection within the previous 6 months.
Study design
In a randomised, double-blind, parallel group design, patients were evaluated on two occasions: before and 24 h after intravenous administration of the TNF-α antagonist, etanercept (10 mg), or saline placebo. A single subcutaneous 25 mg dose of etanercept improves endothelium-dependent vasodilatation within 6 h of administration in patients with heart failure.12 In order to achieve a rapid onset of action, we elected to use the intravenous route, and single intravenous doses of up to 10 mg/m2 have been used safely in patients with heart failure.15 Subcutaneous etanercept has 23% bioavailability and achieves peak plasma concentrations of 0.43 µg/ml at 66 h.16 In contrast, a single intravenous dose of 10 mg etanercept achieves a maximum concentration of 2.32 µg/ml at 50 min.16 Given a plasma half-life of approximately 70 h,16 we judged that 24 h would be an appropriate time point to capture any potential beneficial cardiovascular effects of intravenous etanercept. Randomisation was performed by a computer generated sequence to ensure concealment of treatment allocation and following minimisation for age, sex, time to randomisation, peak troponin, serum cholesterol and cardiovascular risk factors including diabetes mellitus, hypertension and smoking.17 Study procedures were all completed prior to invasive angiography or percutaneous coronary intervention.
Venous sampling and cytokine analysis
Patients abstained from food and caffeine containing drinks for 4 h, and 24 h from ingestion of alcohol prior to phlebotomy. Blood samples were obtained through a 17-gauge cannula with careful attention to ensure smooth extraction of blood to avoid artefactual platelet activation ex vivo, and anticoagulated with D-phenylalanyl-L-propyl-L-arginine chloromethyl-ketone (PPACK). During vasomotor assessments, venous blood was withdrawn simultaneously from each arm and collected into tubes containing acidified buffered citrate (for t-PA assays), trisodium citrate (for plasminogen activator inhibitor type 1 (PAI-1) assays) and potassium EDTA (for cytokine assays). Citrate and acidified buffered citrate samples were centrifuged at 2000 g for 30 min at 4°C and EDTA samples at 1000 g for 10 min at 20°C. Platelet-free plasma was decanted and stored at -80°C before assay. Plasma TNF-α and IL-6 concentrations were determined as described previously18 using ELISA (Quantikine human TNF and IL-6 immunoassays, R&D Systems; and Dako A/S, respectively) and fibrinolytic activities using a photometric method (Coatest t-PA and PAI-1, Chomogenix AB). Haematocrit and white cell count were determined using an automated Coulter counter (Beckman-Coulter).
Vasomotor assessment
Assessment of vasomotor function in response to intra-arterial vasodilators was performed as described previously.19 Briefly, studies were performed in a quiet, temperature-controlled environment with the patient in the supine position throughout. Venous 17-gauge cannulae were inserted into each forearm for blood sampling and a 27-standard wire gauge steel needle placed in the brachial artery of the non-dominant forearm for vasodilator infusions. A baseline 30 min intra-arterial infusion of saline was followed by intra-arterial infusion of substance P at 2, 4 and 8 pmol/min; then acetylcholine at 5, 10 and 20 µg/min; and finally sodium nitroprusside at 2, 4 and 8 μg/min, infused for 10 min at each dose with each agonist separated by a 20 min saline infusion. Forearm blood flow was measured in both arms by venous occlusion plethysmography with the use of mercury-in-silastic strain gauges. Heart rate and blood pressure in the non-infused arm were monitored at intervals throughout with the use of a semiautomated, non-invasive oscillometric sphygmomanometer.
Immunostaining and flow cytometry
All reagents were obtained from AbD Serotec (Oxford, UK) unless otherwise stated. The following antibodies were used: IgG1-FITC, CD14-PE, CD42a-FITC, IgG2a-PE, CD62P-PE, IgG1-PE (eBioscience Ltd. UK) and CD14-RPE and IgG1-RPE (DakoCytomation, Denmark). All antibodies were diluted 1:20 with flow buffer. FACS-Lyse was obtained from Becton-Dickinson (Cowley, UK). Blood anticoagulated with PPACK was labelled with the appropriate antibodies exactly 5 min from collection and incubated in the dark for 20 min at room temperature. Immunolabelling was performed on whole blood to avoid artefactual platelet activation caused by purification procedures. Samples were then fixed and run through a FACSCalibur (Becton-Dickinson) flow cytometer within 2 h of fixing using an established flow protocol.20 For determination of PMA a total of 3000 events were collected in the CD14 monocyte gate. For platelet expression of P-selectin 7500 events were collected in the platelet gate.
Data and statistical analysis
Cytometric data analysis was performed with FlowJo V.7.2.5 (Flow Cytometry Analysis Software). Plethysmographic data and net t-PA release were determined as described previously,19 ,21 as the product of the infused forearm plasma flow (based on the mean haematocrit and the infused FBF) and the concentration difference between the infused ([t-PA] inf) and non-infused ([t-PA] non-inf) arms: estimated net t-PA release=FBF×(1−Hct)×([tPA]inf−[tPA]non-inf). Continuous variables are reported as means±SEM. Analysis of variance with repeated measures and a two-tailed Student t test were performed as appropriate with the use of GraphPad Prism software. The primary outcome measure was PMA. On the basis of previous work examining the effect of clopidogrel on PMA22 we estimated that a 5% absolute change in PMA would be of clinical significance and that at least 12 paired samples would provide an 80% power to detect a 10% absolute difference in PMA from baseline at a significance level of 5%.
Results
26 patients hospitalised with type 1 acute non-ST elevation myocardial infarction were recruited into the study. Patients were well matched with respect to all relevant baseline characteristics (table 1). No additional medical therapy was instituted during the study period. No serious adverse events occurred during the study.
Baseline demographics and medical therapy of study population
Inflammatory response and cytokine analyses
Consistent with effective conjugation of circulating TNF-α, plasma TNF-α concentrations increased in all patients following etanercept infusion (254±15 vs 0.12±0.02 pg/ml; p<0.0001). At 24 h, treatment with etanercept was associated with a reduced neutrophil count (8.8±0.6 vs 7.4±0.5 cells 109/l; p=0.02), and a rise in the lymphocyte count (2.3±0.2 vs 2.7±0.26; p=0.001), with a reduction in the neutrophil to lymphocyte ratio following etanercept compared with placebo (−1.3±0.4 vs 0.17±0.2; p=0.001). Plasma interleukin-6 concentrations were similarly reduced (10.6±4.0 vs 5.8±2.0 pg/ml; p=0.01). No significant differences were observed at 24 h compared with baseline in those patients randomised to placebo (p>0.05 for all; table 2).
Inflammatory response, indices of platelet activation and fibrinolytic function
Platelet activation
PMA and platelet P-selectin expression were similar between the groups at baseline. Following etanercept infusion, there was a 50% relative increase in PMA (30±5 vs 20±3%; p=0.02) compared with baseline. PMA was however unaffected by saline placebo infusion (28±5 vs 33±6%; p=0.23). Platelet P-selectin expression was not affected by either treatment (p>0.05 for both; table 2).
Vasomotor response
Of the 26 patients enrolled, 15 underwent vascular assessment (eight randomised to receive etanercept). Heart rate and systemic blood pressure were similar in both groups and were unaffected by either treatment (data not shown). Baseline forearm blood flow in the non-infused arm was similar and was unaffected by either treatment. There was a dose-dependent increase in forearm blood flow with all intra-arterial vasodilators: substance P, acetylcholine and sodium nitroprusside (p<0.001). However, there were no differences in the dose–response curves between etanercept and placebo (p>0.1 for all; figure 1).

Peripheral vasomotor assessment. Forearm blood flow in response to incremental doses of sodium nitroprusside, substance P and acetylcholine in patients with acute myocardial infarction 24 h following etanercept compared with placebo. Forearm blood flow increases in response to vasoactive infusion (ANOVA<0.0001 for all); however, there was no difference between treatments (ANOVA≥0.2; ≥0.7 for interaction for all) or at 24 h compared with baseline (ANOVA>0.1).
Fibrinolytic response
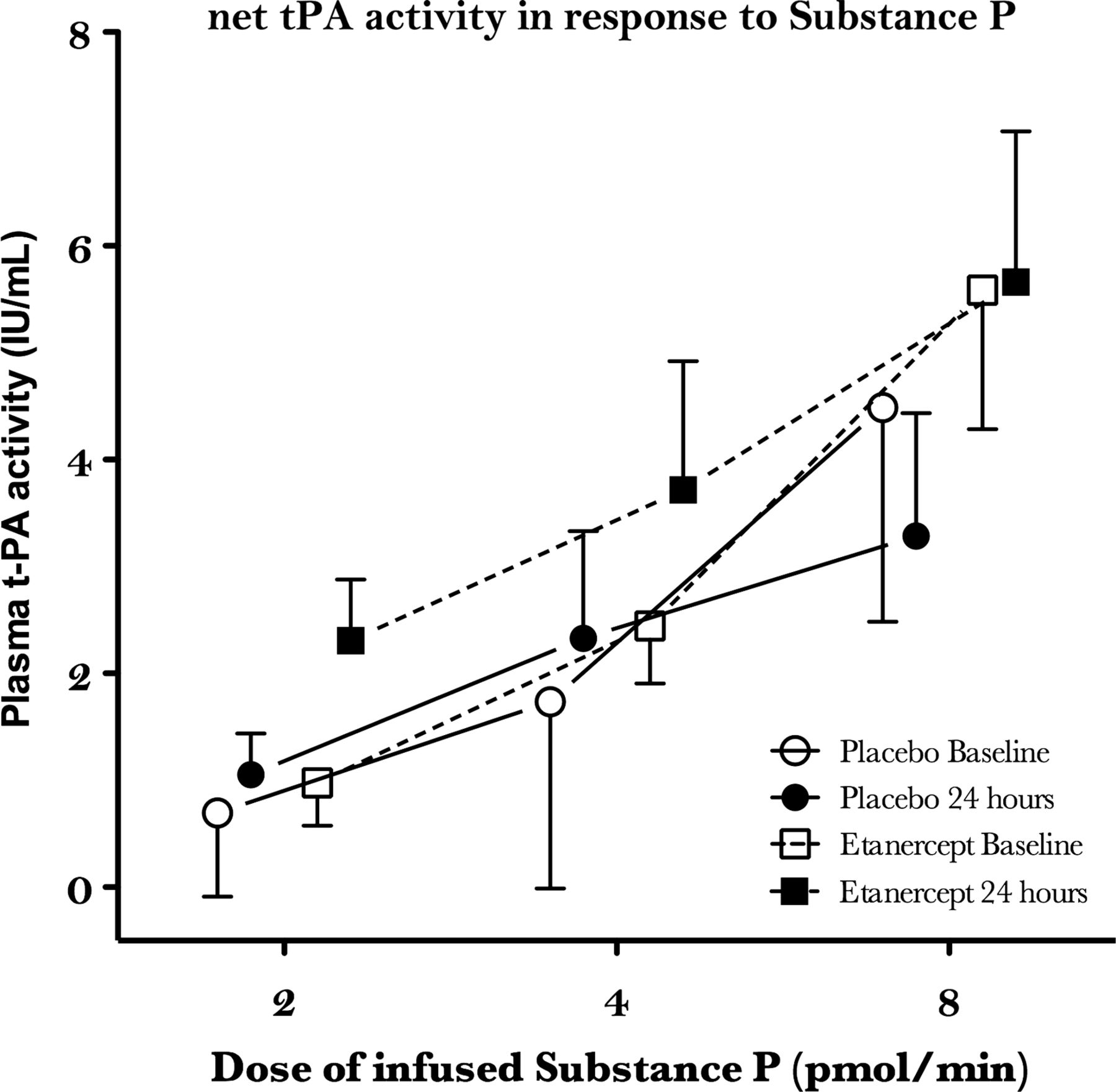
Plasma t-PA activity concentrations were similar between groups at baseline and were unaffected by saline placebo (p=1.0). Following etanercept, plasma t-PA activity concentrations were reduced at 24 h (0.8±0.1 vs 0.5±0.1 IU/ml; p=0.001). Compared with the non-infused arm, substance P caused dose-dependent increases in plasma t-PA activity (ANOVA: p=0.001). However, dose–response curves were similar at 24 h and were unaffected by either treatment (p>0.1 for all; table 2; figure 2). Plasma PAI-1 antigen concentrations were similar between the groups at baseline and did not differ following either treatment (p>0.1).

{kind=link}
{kind=link}
Net t-PA activity in response to infusion of substance P. Net t-PA is calculated as the difference between the infused and non-infused arm corrected for haematocrit. Net t-PA activity increased in response to t-PA infusion (ANOVA<0.01); however, there were no differences between treatments (ANOVA>0.05 for all) or at 24 h compared with baseline (ANOVA>0.1).
Discussion
This is the first study to evaluate the effect of TNF-α antagonism in patients with AMI. We demonstrate that although providing a modest anti-inflammatory effect, treatment with etanercept was not associated with any improvement in peripheral vasomotor or fibrinolytic function. Of potential concern, we identified an increase in platelet monocytes aggregation signalling a potentially adverse pro-thrombotic effect. We conclude that TNF-α antagonism does not appear to provide beneficial cardiovascular effects in patients with AMI and is unlikely to be a beneficial novel therapy in this setting.
Etanercept is a dimeric fusion protein that consists of the extracellular ligand-binding domain of the human TNF receptor, linked to the Fc portion of human IgG1. Etanercept inactivates circulating TNF-α by binding to it, thereby preventing activation of endogenous TNF-α receptors. Consistent with previous reports,23 we observed a dramatic increase in plasma TNF-α concentrations following treatment with etanercept. Etanercept paradoxically increases the immunoreactivity and stability of circulating TNF-α, probably as a consequence of its dimeric structure, which can cause an increase in the relative quantities of circulating homotrimeric, as opposed to monomeric or dimeric, TNF-α.24 This paradoxical effect of TNF-α antagonism can lead to an effective increase in TNF-α mediated cytotoxicity, and this effect appears to be particularly pronounced where there is a marked molar excess of TNF-α relative to etanercept. Although we did not observe any direct vascular effects associated with the increase in plasma TNF-α concentrations, we did observe a 50% relative increase in PMA, a measure of platelet activation. We have previously observed an approximate 20% fall in PMA following treatment with clopidogrel,22 suggesting that the increase associated with treatment with etanercept is clinically relevant. These findings are consistent with previous in vitro studies showing that TNF-α can cause an increase in platelet aggregation in patients with heart failure, probably through activation of the arachadonic acid pathway.25 ,26 Enhanced platelet activation through this mechanism may in part explain the thrombotic tendency associated with disorders characterised by high plasma concentrations of TNF-α.7 However, we did not detect a difference in P-selectin expression, an alternate measure of platelet activation. P-selectin expression is generally very transient with low levels of expression, making this a less reliable in vivo marker of platelet activation.27 Although this may explain our discrepant findings, we would suggest caution in interpreting the significance of our findings with PMA given that this was a single measure of platelet activation.
The recognition that heart failure is characterised by high plasma concentrations of TNF-α has stimulated intensive research of TNF-α antagonism in these patients. Interestingly, although it has been established that antagonism of TNF-α in the setting of congestive heart failure can ameliorate endothelial dysfunction,12 and improve left ventricular systolic dysfunction,28 several large clinical trials of TNF-α antagonists in patients with congestive heart failure have failed to demonstrate any clinical benefits associated with this form of treatment.29 Furthermore, there is concern over the safety of TNF-α antagonism in these patients, as these trials suggest that there may be an increased risk of the development of worsening heart failure in those patients receiving higher doses of TNF-α antagonists.30 The present study suggests that increased platelet activation may in part explain these observations, as any beneficial endothelial effects associated with etanercept may be offset by an increase in thrombogenicity. Patients with coronary artery disease generally have lower plasma concentrations of TNF-α than patients who have progressed to develop heart failure, limiting extrapolation between these two patient populations.
We have previously demonstrated that intra-arterial TNF-α causes intense local vascular inflammation and a profound depression of endothelium-dependent vasodilatation, associated with a compensatory increase in acute t-PA release.31 We therefore expected that antagonism of TNF-α would ameliorate vascular inflammation, and lead to an improvement in vasomotor function commensurate with a fall in plasma t-PA concentrations. Consistent with this hypothesis, we found that basal t-PA activity concentrations were diminished following etanercept. We have previously hypothesised that t-PA concentrations are increased in response to vascular inflammation as a compensatory mechanism.31 The reduction in basal t-PA activity following etanercept may therefore reflect a fall in basal t-PA release through amelioration of the systemic inflammatory response associated with AMI. However, neither PAI-1 activity nor net t-PA release in response to substance P were affected by etanercept infusion, suggesting that the magnitude of this effect was modest. Furthermore, we saw no effect on vasomotor function. We suggest that the lack of effect on the vasomotor response may have been due to the relatively low plasma concentrations of TNF-α in this cohort of patients. Although concentrations are increased in patients with AMI,8 ,9 patients in the present study had low plasma TNF-α concentrations. The reason for this is unclear; however, it may be explained by the timing of patient recruitment into the study (on average 3 days following presentation) as TNF-α plasma concentrations are maximal within the first 24 h following AMI, falling off markedly thereafter, particularly in the absence of heart failure.32 Selection of patients with relatively higher plasma concentrations of TNF-α may have increased the likelihood of observing improvements in vascular function.
Study limitations
Although appropriately powered for platelet monocyte aggregation, confounding factors such as those related to pharmacokinetic and pharmacodynamic variation between subjects and the effects of concurrent medical therapy are challenging to standardise. To corroborate the observed increase in PMA associated with etanercept, further measures of platelet function such as platelet aggregation would be useful.
We cannot exclude the possibility that more rapid treatment (within the first 24 h) and higher doses of etanercept may have produced different results. However, the marked increase in circulating TNF-α, commensurate with a reduction in systemic inflammation following administration of etanercept, gives us reassurance that we exerted a meaningful biological effect, and that the absence of any alteration in vascular function following treatment was not as a result of a type-2 error.
Conclusions
The administration of a single dose of etanercept following AMI provides a modest anti-inflammatory effect associated with a mild reduction in basal plasma t-PA activity. However, this does not translate into improvements in peripheral vasomotor function or acute t-PA release. Of concern, etanercept potentiates platelet–monocyte aggregation, suggesting an adverse effect of TNF-α antagonism in patients with acute coronary syndromes through enhanced platelet activation. Further research is required to explore the effects of etanercept on platelet activation and function as such effects may in part explain the possible deleterious effects observed in clinical trials of TNF-α antagonism in other cardiovascular disorders.
Acknowledgments
All authors contributed significantly to the work. The research was devised by SAH, SC and DEN, and conducted by GJP, JND, AJL, NLMC, SDR, NLM and EK. GJP analysed the data and wrote the manuscript with input from the coauthors. GJP takes overall responsibility for the manuscript.
References
Footnotes
-
Funding The research was funded through the University of Edinburgh with additional support from the British Heart Foundation (PG/2001068) who also supported Dr Padfield (SS/CH/92010 and PG/07/ 012) and Professor Newby (CH/09/002) to undertake the work. The Wellcome Trust Clinical Research Facility is supported by NHS Research Scotland through NHS Lothian. All authors have read and approved the manuscript for submission. The paper is not under consideration elsewhere, none of the paper's contents have been previously published. Technical appendix, statistical code and dataset are available on request.
-
Competing interests None.
-
Ethics approval LREC/2001/4/18.
-
Provenance and peer review Not commissioned; externally peer reviewed.