Article Text

Abstract
A 35 year old woman presented with acute myocardial infarction without any of the usual risk factors: she had never smoked; she had normal blood pressure; she did not have diabetes; plasma concentrations of total cholesterol and high and low density lipoprotein cholesterol, fibrinogen, homocysteine, and Lp(a) lipoprotein were normal. She was not taking oral contraceptives or any other medication. Coronary angiography showed occlusion of the left anterior descending coronary artery but no evidence of arteriosclerosis. Medical history disclosed a previous leg vein thrombosis with pulmonary embolism. Coagulation analysis revealed protein C deficiency. The recognition of protein C deficiency as a risk factor for myocardial infarction is important as anticoagulation prevents further thrombotic events, whereas inhibitors of platelet aggregation are ineffective.
- myocardial infarction
- risk factors
- protein C deficiency
- anticoagulation
Statistics from Altmetric.com
A small minority of patients with myocardial infarction have none of the usual and well defined risk factors. We report a 35 year old woman with recurring myocardial infarction in whom recognition of a coagulation defect led to specific preventive measures.
Case report
In March 1996, a 35 year old woman (168 cm, 65 kg) was admitted with an anterior wall myocardial infarction complicated by left sided cardiac failure. The patient had never smoked, she did not have diabetes mellitus or hypertension. She was not taking oral contraceptives or any other medication. In 1985, she had a deep leg vein thrombosis followed by pulmonary embolism. She was treated with phenprocoumon for one year. In February 1996 she experienced a transitory paralysis of the left leg. She did not seek medical attention for this condition. Family history revealed that the patient’s father had died of pulmonary embolism, her sister had a calf vein thrombosis, and her mother was healthy.
An ECG showed a loss of R in the anterior myocardial wall (V1–6). Transthoracic echocardiography showed highly reduced contractibility (ejection fraction ∼ 20%) as well as dilatation and akinesia of the apical and septal parts of the left ventricle. No thrombolytic treatment was provided as the infarction had occurred several days earlier. Coronary angiography showed occlusion of the left anterior descending artery but no coronary arteriosclerosis (fig 1). The next day, despite effective anticoagulation (partial thromboplastin time 62 seconds) with unfractionated heparin (30 000 U/day), the patient had another anterior myocardial wall infarction. Lysis with tissue-type plasminogen activator did not lead to recanalisation. The course was complicated by repeated ventricular flutter requiring cardioversion on several occasions. The patient was mobilised with amiodarone, captopril, digoxin, frusemide, and atenolol treatment.
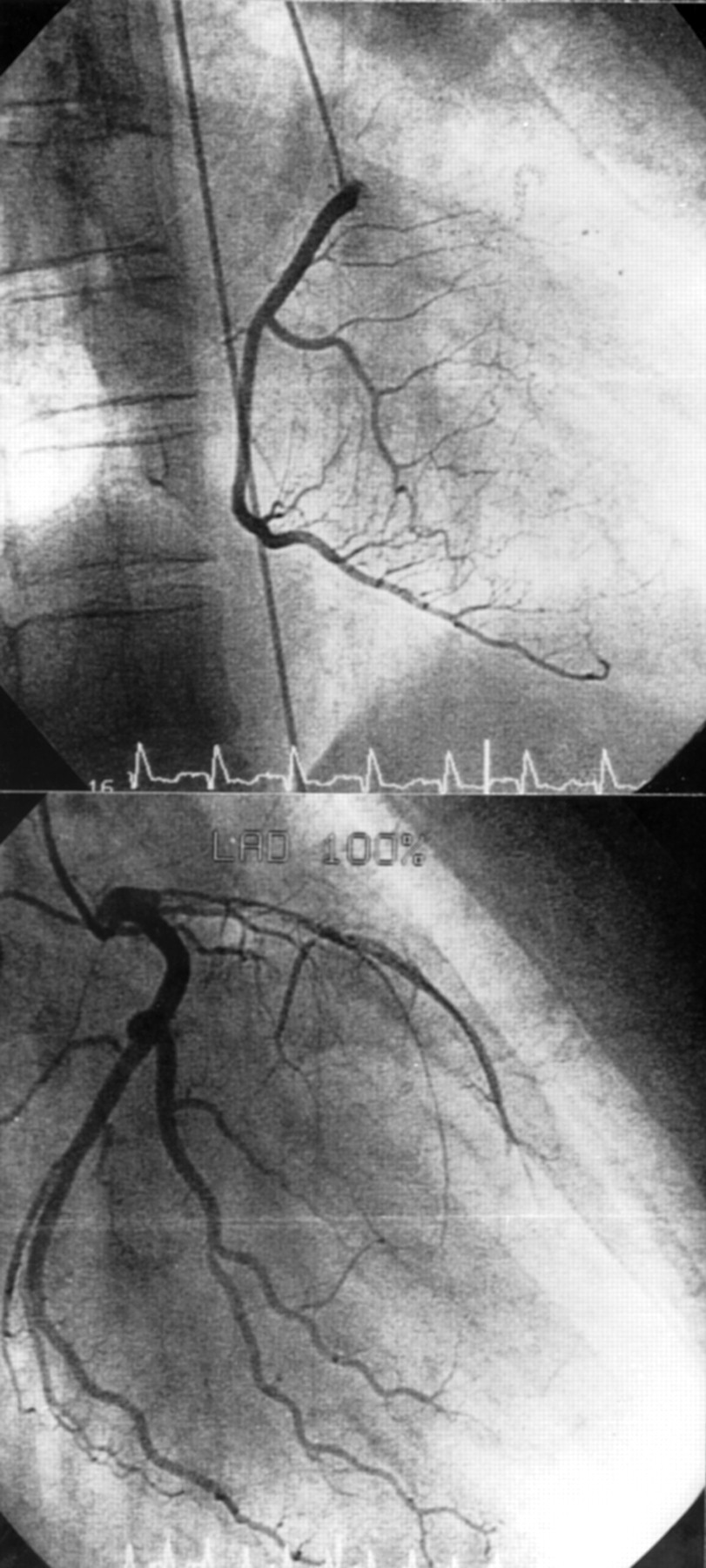
{kind=link}
Angiogram of the left (top) and the right (bottom) coronary artery reveals no arteriosclerosis but complete occlusion of the left anterior descending artery.
Coagulability testing showed decreased protein C activity of 57% (normal value > 70%). Cholesterol (high density lipoprotein (HDL) 1.11 mmol/l, low density lipoprotein (LDL) 1.73 mmol/l), fibrinogen, homocysteine, Lp(a) lipoprotein, protein S, antithrombin-III, and activated protein C resistance were normal. The patient was anticoagulated (INR 3.0–4.0) with phenprocoumon. At follow up examination in March 1997 she was physically active in the house and feeling well.
Discussion
Protein C, a serine protease activated by the thrombin–thrombomodulin complex, is part of the inhibitor system of plasma coagulation. Activated protein C exerts a negative feedback on the intrinsic and extrinsic pathways through proteolytic inactivation of factors Va and VIIIa in the presence of protein S and phospholipids. It increases fibrinolytic activity, possibly by neutralisation of the plasminogen activator inhibitor 1; therefore, deficiency of protein C induces hypercoagulability. The genetic defect is a single point mutation in exon 7 of the protein C gene located on chromosome 2q13–q14.1 Protein C deficiency usually manifests as thrombosis of the venous system. The prevalence of arterial thrombosis in 337 heterozygotes was 7.1%.2 It has been suggested that additional vascular risk factors are required for the involvement of the arterial system.3 A medline search revealed three detailed publications on patients with myocardial infarction associated with protein C deficiency,2-4 but these patient had one or more other risk factors (smoking, diabetes mellitus, abnormal concentrations of HDL, LDL, fibrinogen, Lp(a) lipoprotein, or homocysteine) In contrast to other congenital risk factors, there is an effective treatment for protein C deficiency. Whereas thrombocyte aggregation inhibitors such as aspirin and ticlopidine are ineffective, anticoagulation with coumarins prevents further thrombotic events. Our case refutes the concept that there is not a useful role for measurement of endogenous anticoagulant pathways in assessing patients at risk for arterial thrombosis.5 A complete medical history is crucial for an accurate diagnosis and effective prevention of further thrombotic events.