Article Text

Abstract
BACKGROUND Two common mutations of the haemochromatosis associated gene (HFE) (cys282tyr (C282Y) and his63asp (H63D)) have been implicated in haemochromatosis and as modulators in cardiovascular disease.
OBJECTIVE To investigate the role of these mutations in the pathogenesis of idiopathic dilated cardiomyopathy.
DESIGN AND SETTING Case–control and prospective cohort study of patients attending a cardiomyopathy unit in a tertiary referral cardiac centre.
METHODS 207 unrelated white patients with dilated cardiomyopathy, followed up for 259 patient years, and 200 controls were tested for HFE C282Y and H63D mutations by polymerase chain reaction and restriction digestion.
RESULTS 31/207 patients (15%)v 24/200 controls (12%) carried C282Y (adjusted odds ratio (OR) 1.2 (95% confidence interval 0.7 to 2.2)), 74/207 (36%) v 53/200 (27%) carried H63D (OR 1.6 (1.1 to 2.5)), and 10/207 (4.8%) v4/200 (2%) were compound heterozygotes (OR 2.6 (0.8 to 8.5)). Four patients and six controls were H63D homozygous and one was C282Y homozygous. There was a progressive increase in mean serum iron ([Fe]) and transferrin saturations from patients with no mutation ([Fe] = 16.3 μmol/l, transferrin saturation = 23.7%) through H63D heterozygotes (17.5 μmol/l, 25.8%), C282Y heterozygotes (17.1 μmol/l, 26.6%), H63D homozygotes (20.0 μmol/l, 33.5%), compound heterozygotes (26.8 μmol/l, 41.7%), and C282Y homozygotes (34 μmol/l, 71%). At follow up (median 90 months) the rate of death or cardiac transplantation was 52/207 (25%). C282Y heterozygotes had less ventricular dilatation (mean (SD): 59.9 (1.7) mmv 64.9 (0.9) mm, p < 0.05), better fractional shortening (24 (1.7)% v 18.8 (1.4)%, p < 0.01), and a trend towards improved survival without transplantation. [Fe] and transferrin saturation did not correlate with disease severity and were not associated with reduced survival.
CONCLUSIONS The frequency of the H63D mutation is significantly increased in patients with idiopathic dilated cardiomyopathy. As H63D has a relatively minor effect on iron status, the mechanism of this association may be unrelated to iron metabolism.
- dilated cardiomyopathy
- genetics
- haemochromatosis
Statistics from Altmetric.com
Idiopathic dilated cardiomyopathy is a myocardial disease with an annual incidence of 5–8/100 000 and a prevalence of 40/100 000.1 It accounts for up to 50% of indications for cardiac transplantation2 and up to 20% of patients in large scale heart failure trials.3 Approximately 50% of cases are familial, and the most frequent inheritance pattern is autosomal dominant.4 Several autosomal dominant disease loci have been identified,5-8 and genes identified as causing autosomal dominant dilated cardiomyopathy include actin,9desmin,10 and lamin a/c.11 Families vary from those with a high degree of penetrance, consistent with the presence of a single gene disease causing mutation, to those with relatively few individuals affected, suggesting the presence of a genetic predisposition rather than a direct disease causing mutation.
Several potential disease modifying or predisposing genes have been investigated. Associations have been reported with angiotensin converting enzyme genotypes12 and with HLA subtypes.13 The recently described hereditary haemochromatosis gene on chromosome 6p (termed the HFE gene) is a candidate as a disease modifying gene in dilated cardiomyopathy.14 Two mutations of this gene are associated with the development of hereditary haemochromatosis: the first, leading to a substitution of tyrosine for cysteine (cys282tyr, C282Y), causes hereditary haemochromatosis in homozygotes; the second, leading to a substitution of aspartate for histidine (his63asp, H63D), may cause hereditary haemochromatosis when associated with C282Y (compound heterozygotes).14-17 These are common mutations in white populations of North European extraction, in whom 12% are heterozygous for C282Y and 24% heterozygous for H63D.18 ,19 The high carrier rates may be accounted for by heterozygote advantage with regard to iron conservation. As left ventricular dilatation and heart failure are features of hereditary haemochromatosis, we postulated that HFE mutations may be involved in the aetiology of dilated cardiomyopathy or be disease modifying factors.
Our aim in this study was to determine the frequency of HFE mutations among white patients with dilated cardiomyopathy relative to representative controls.
Methods
PATIENTS
Patients were 207 consecutive unrelated white index cases evaluated in one centre over a six year period and diagnosed as having dilated cardiomyopathy by World Health Organization criteria.20 At initial assessment, blood was stored at −80°C in a tissue library for future genetic analysis, in accordance with local research ethics committee approval and with informed consent. Evaluation included clinical history, physical examination, ECG, cross sectional echocardiography, symptom limited maximum exercise test with respiratory gas sampling on a Sensormedics ergometrics 800 S cycle ergometer (Bitz, Germany), and 24 hour ECG monitoring (Marquette Electronics, Toulouse, France). Coronary angiography was performed in patients over 40 years or when clinically indicated. Subjects were reviewed at least annually until death or transplantation. Subjects with ⩾ 50% obstruction of one or more coronary arteries, active myocarditis, specific primary or secondary heart muscle disease, sustained systemic arterial hypertension, isolated right ventricular dilatation, and valvar or pericardial disease were excluded. For the purposes of identifying familial disease, detailed pedigrees were constructed and asymptomatic first and second degree relatives of index cases were systematically evaluated as above. If disease was identified in family members, the pedigrees were further evaluated. Index cases were classified as having familial disease according to criteria recently proposed by the collaborative research group committee of the European human and capital mobility project on familial dilated cardiomyopathy.21
CONTROLS
Blood from 141 healthy white blood donors and 59 white factory workers was used for control assays. Age and sex distribution of controls was recorded to enable correction for these factors in statistical comparisons with cases.
HFE GENE MUTATION ASSAYS
The methodology for the detection of HFE mutations has been described previously.14 Briefly, the regions of HFE containing the C282Y and H63D mutations were amplified by polymerase chain reaction (PCR) and then subjected to restriction enzyme analysis. The primers were: H63DF-5′ ACA TGG TTA AGG CCT GTT GC; H63DR-5′ GCC ACA TCT GGC TTG AAA TT for the H63D locus and C282YF-TGG CAA GGG TAA ACA GAT CC; C282YR-5′ 5′CTC AGG CAC TCC TCT CAA CC for the C282Y locus. Amplification was performed using 25 μl reaction mixtures containing 10 mM Tris-HCl, pH 8.3; 50 mM KCl; 1.5 mM MgCl2; 0.2 mM of each dNTP; 0.5 μM of each primer; 1 U of Amplitaq Gold DNA polymerase (Perkin Elmer); and 100 ng of genomic DNA. The enzyme was heat activated at 94°C for 10 minutes, followed by 35 cycles of denaturation at 94°C for one minute, annealing at 59°C for one minute, and extension at 72°C for one minute, with digestion of products after a final extension period of three minutes at 72°C. Digestion was carried out with Sau3A, a recognition site for which is eliminated by the H63D mutation, and RsaI, which recognises a site created by the C282Y mutation.
BIOCHEMICAL ASSAY
Each patient underwent evaluation of serum urea and creatinine, liver function, iron, ferritin, and transferrin saturations. Ferritin was assayed by a solid phase, two site fluoroimmunometric assay (Autodelphia, Wallac, Turku, Finland), and iron was assayed using a thin film incorporated on a dry slide (Kodak Ektachem, Johnson and Johnson, Rochester, New York, USA). Total iron binding capacity was measured in serum by adding excess ferric iron to saturate all transferrin binding sites, removing unbound iron with alumina and measuring iron as above (Technicon RA1000 protocol, July 1998).
STATISTICAL ANALYSIS
The odds ratio (OR) and 95% confidence interval (95% CI) of dilated cardiomyopathy associated with the presence of each variant allele were calculated using unconditional logistic regression, adjusting for sex and age. The effect of genotype on iron concentrations and transferrin saturations was determined using analysis of covariance, adjusting for age and sex, comparing each genotype with non-carriers of either mutation. A formal test for skewness was made using the Kolmogorov–Smirnoff test. The influence of genotype and iron levels on clinical variables was analysed using Kaplan–Meier survival analysis (survival without cardiac transplantation) and Cox regression.
The power of the study was computed as the probability of detecting an association between C282Y/H63D variants and dilated cardiomyopathy at the 0.05 level of significance assuming a genotypic risk of 3.0, with an additional calculation to determine the ability to detect differences between HFE genotypes with regard to transferrin saturations based on available data, again at the 0.05 level of significance. Statistical analyses were undertaken using the software packages STATA (version 5.0, STATA Corporation, Texas, USA) and SPSS for windows (version 7.0, SPSS Inc, Chicago, Illinois, USA).
Results
FREQUENCY OF MUTATIONS
Table 1 shows the frequency distribution of the C282Y and H63D genotypes among patients with dilated cardiomyopathy and controls. Both populations were in Hardy–Weinberg equilibrium for C282Y and H63D. Table 2 lists frequencies and odds ratios for heterozygotes for either mutation, carriers of any mutation, and homozygotes or compound heterozygotes in controls, all patients with dilated cardiomyopathy, and male subjects as a separate group. Based on the carrier frequency of C282Y and H63Y, this study has 80% power to detect a 2.2-fold and a 1.9-fold increase in risk, respectively. The variant allele C282Y was present in 21 cases (15%) and H63D in 56 cases (39%) (allele frequencies 0.08 and 0.20, respectively); C282Y was present in 24 controls (12%) and H63D in 53 controls (27%) (allele frequencies 0.05 and 0.14, respectively).
Distribution of the C282Y and H63D mutation among subjects with dilated cardiomyopathy (DCM) and controls
Prevalence of mutations among controls, all patients with dilated cardiomyopathy (DCM), and patients with familial and sporadic disease
The frequency of the C282Y mutation was not increased in patients with dilated cardiomyopathy (OR = 1.2; 95% CI, 0.7 to 2.2). In contrast, the frequency of H63D was significantly higher in the patients with dilated cardiomyopathy (OR = 1.7; 95% CI, 1.1 to 2.5). These odds ratios were not altered by analysing familial or sporadic disease separately or by analysing only male patients (H63D, OR = 1.7 (95% CI, 1.0 to 2.8, p < 0.05)).
HAEMOCHROMATOSIS MUTATIONS AND CLINICAL FEATURES OF DILATED CARDIOMYOPATHY
The influence of the C282Y and H63D genotypes on the clinical characteristics and outcome in the patients with dilated cardiomyopathy is shown in table 3.
Clinical features of patients with dilated cardiomyopathy according to genotype
Carriers of C282Y had a shorter duration of illness before presentation, less severe ventricular dilatation (mean (SD): 59.9 (1.7) mm v 64.9 (0.9) mm, p < 0.05), and better fractional shortening (24 (1.7)%v 18.8 (1.4)%, p < 0.01). There was no difference between carriers and non-carriers regarding severity of symptoms, consumption of ethanol, presence of familial disease, presence of arrhythmia, or rate of death or cardiac transplantation.
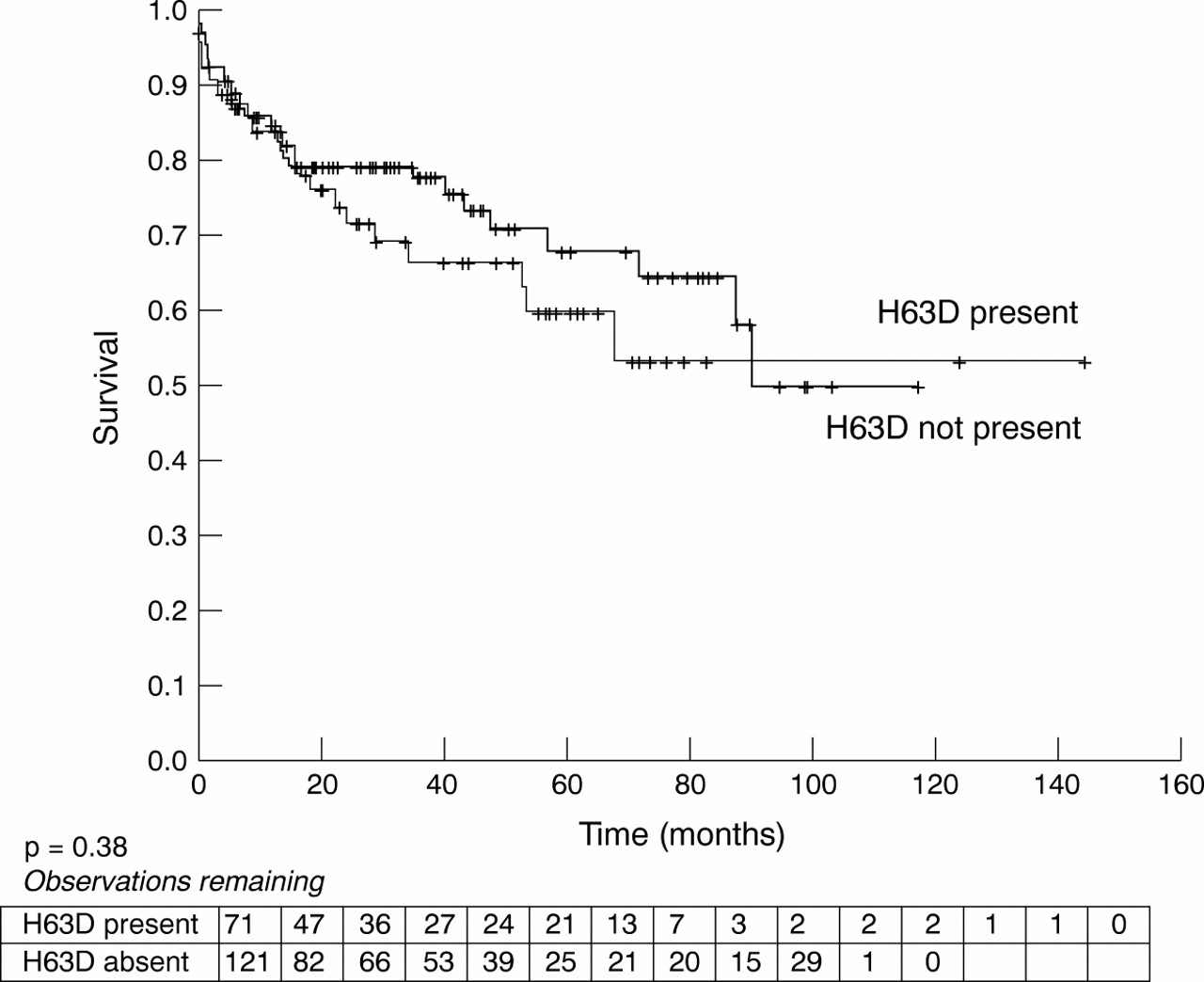
For the H63D genotype there was no difference between carriers and non-carriers with regard to any clinical outcome.
Figures 1 and 2 shows death and transplant-free survival rates in patients with and without C282Y mutations (fig 1) and H63D mutations (fig 2) for a median follow up of 90 months. There was a non-significant trend towards improved survival without cardiac transplantation among carriers of C282Y. No survival difference was detected between patients with and without the H63D mutation.

Death or transplant-free survival of patients carrying the C282Y variant allele.

{kind=link}
{kind=link}
Death or transplant-free survival of patients carrying the H63D variant allele.
INFLUENCE OF MUTATIONS ON INDICES OF IRON METABOLISM
Using actual sample sizes, the differences in transferrin saturations between each group and the normal group that could be detected with a power of 0.8 were 4.5% for H63D heterozygotes, 6.9% for C282Y heterozygotes, 14.2% for H63D homozygotes, 9.2% for compound heterozygotes, and 28% for C282Y homozygotes.
Observed indices of iron metabolism as well as renal function, liver function, and glucose according to genotype are shown in table 4. There were no differences in liver function, renal function, glucose, or haemoglobin concentrations between any group. There was a progressive increase in mean serum iron concentrations and transferrin saturations from patients with no mutation ([Fe] = 16.3 μmol/l, transferrin saturation = 23.7%) through patients heterozygous for the H63D variant ([Fe] = 17.5 μmol/l, transferrin saturation = 25.8%), heterozygous for the C282Y variant ([Fe] = 17.1 μmol/l, transferrin saturation = 26.6%), patients homozygous for H63D([Fe] = 20.0 μmol/l, transferrin saturation = 33.5%), compound heterozygotes ([Fe] = 26.8 μmol/l, transferrin saturation = 41.7%) and patients homozygous for C282Y ([Fe] = 34 μmol/l, transferrin saturation = 71%). After controlling for age and sex differences serum iron concentrations and transferrin saturation were significantly higher in compound heterozygotes (p < 0.01) and in C282Y homozygotes (p < 0.01) than in patients with no mutation.
Biochemical indices and iron concentrations according to genotype
CLINICAL FEATURES OF COMPOUND HETEROZYGOTES
Compound heterozygote patients had transferrin saturation levels that were significantly higher than heterozygotes but below 50%, in contrast with the majority of patients with iron storage disease who have transferrin saturation of more than 50%.22 Ten of these 11 patients were male, and the sole woman was postmenopausal. In this group there was no biochemical evidence of liver disease or diabetes (table 4) and there were no clinical differences between these patients and the remaining patients with dilated cardiomyopathy (table5). They were followed for a median of 30 months (range 0–72 months). During that time there was no evidence of progressive cardiomyopathy or of the development of liver disease, diabetes, or other clinical features of haemochromatosis. In one patient who had undergone transplantation shortly after enrolment there was no histological evidence of iron deposition in the explanted heart.
Clinical features of compound heterozygote
INFLUENCE OF IRON CONCENTRATIONS ON CLINICAL OUTCOMES IN DILATED CARDIOMYOPATHY
There were no correlations between iron concentrations, ferritin concentrations, or transferrin saturations and left ventricular dimensions, systolic function, functional class, or peak oxygen consumption (Vo 2). In order to determine whether iron status had an influence on survival in dilated cardiomyopathy, univariate and multivariate predictors of survival were determined (table 6). Factors associated with a poor prognosis were: increased serum creatinine, reduced systolic function, increased left ventricular end diastolic dimension, higher New York Heart Association (NYHA) functional class, low peak Vo 2, and left bundle branch block on the ECG. Transferrin saturations were higher in survivors than in non-survivors (27.3 (11.2)%v 20.0 (11.0)%, p < 0.01). Other indices of iron metabolism did not differ between survivors and non-survivors. Following Cox regression analysis in which the model included age, sex, left ventricular diastolic dimension (LVDD), fractional shortening, bundle branch block, NYHA class, peak Vo 2, transferrin saturation, and creatinine, only LVDD (OR (per mm increase) = 1.07 (1.00 to 1.15)) and peak Vo 2(OR (per ml decrease) = 1.32 (1.11 to 1.56)) were independently associated with worse survival.
Predictors of survival from death or transplantation in patients with dilated cardiomyopathy
Discussion
Genetic polymorphisms such as of the renin-angiotensin system23-25 are recognised modulators of cardiovascular biology. The potential of the C282Y and H63D mutations to modify expression of diseases other than haemochromatosis has been demonstrated in porphyria cutanea tarda, in which iron contributes to reversible inactivation of uroporphyrinogen decarboxylase. Depletion of hepatic iron stores leads to reversal of enzyme inhibition and clinical remission. Four studies involving different populations of European descent have shown significantly increased frequencies of the C282Y mutation in sporadic porphyria,26-29 while one study, involving Italian patients, showed an increased frequency of H63D (not associated with increased iron indices), but not of C282Y.30 More recently, HFE C282Y has been associated with an increased risk of first acute myocardial infarction among men31 and total cardiovascular mortality among women. The mechanism is also thought to be related to iron metabolism, as iron depletion reduces myocardial injury and atherogenesis.32Our present study explored the role of haemochromatosis mutations in dilated cardiomyopathy.
HEREDITARY HAEMOCHROMATOSIS MUTATIONS IN DILATED CARDIOMYOPATHY: ROLE OF IRON OR OTHER MECHANISMS
We found no increase in the frequency of C282Y in patients with dilated cardiomyopathy, in whom the frequency was 15%, similar to controls and to the UK population.33 In contrast, H63D was found with increased frequency in dilated cardiomyopathy, suggesting that it may predispose to this disease. However, our data and data from large population studies18 suggest that H63D appears to have little effect on iron metabolism compared with C282Y. Therefore, although iron may predispose to myocyte damage by various mechanisms including iron catalysed lipid peroxidation34 and increased susceptibility to toxins such as ethanol and to viral infections,34 ,35 the predominance of H63D rather than C282Y suggests that a different mechanism may be involved.
Other observations supporting an alternative mechanism are as follows. First, analysis of male patients only, in whom transferrin saturation was higher, did not strengthen the association of either mutation with dilated cardiomyopathy. Second, as discussed below, in compound heterozygotes in whom there was notably increased transferrin saturation there was no evidence of progressive iron related disease and no evidence of myocardial iron deposition. Third, there was no increase in severity of disease or reduction in survival in patients carrying mutations which increased transferrin saturation, and there was no correlation between serum iron concentrations and severity of disease. Finally, in other conditions such as porphyria cutanea tarda and atherosclerosis, in which HFE mutations are thought play a role through iron related mechanisms, the mutation implicated is, with one exception,30 C282Y and not H63D.
An alternative explanation of the association is suggested by the location of the HFE gene in close proximity to the human leucocyte antigen (HLA) locus. Consistent with data linking dilated cardiomyopathy with autoimmunity,36 ,37 H63D may predispose to dilated cardiomyopathy through immune mechanisms, determined either by the gene product or by the product of a gene with which it may be in linkage disequilibrium. Evidence of immunological abnormalities including abnormal CD8+ T cell counts38 ,39in haemochromatosis supports this hypothesis. Coupled with these observations, our data support the merits of undertaking further studies exploring immunological indices in patients with dilated cardiomyopathy who have HFE H63D.
DOES A SUBSET OF PATIENTS HAVE SUBCLINICAL HEREDITARY HAEMOCHROMATOSIS?
Both C282Y homozygosity and compound heterozygosity are associated with clinical hereditary haemochromatosis.16 The observation of an increased frequency of C282Y homozygotes/compound heterozygotes among male patients with dilated cardiomyopathy is consistent with the possibility that asubset of these patients has subclinical hereditary haemochromatosis and may benefit from iron load reduction by venesection or iron chelation. This possibility is supported by the fact that the increased frequency of C282Y was only significant in the male subgroup, which one would expect to be most affected by the mutations, and by the observation that the one female compound heterozygote in the cohort was postmenopausal. However, close inspection of the compound heterozygote group yields no evidence of hereditary haemochromatosis in these patients. Despite follow up for a median of 30 months, they did not develop evidence of liver disease, diabetes, or other clinical features of hereditary haemochromatosis. There was also no deterioration in cardiac function, which might be expected if there was progressive untreated iron overload. One compound heterozygote, who underwent cardiac transplantation, had no evidence of iron deposition in his myocardium. These observations suggest that the presence of both mutations does not cause dilated cardiomyopathy through subclinical hereditary haemochromatosis in this population, and the increased frequency of compound heterozygotes may simply reflect the increased frequency of the individual mutations, particularly H63D, in the population. However, longer follow up may be required for definitive exclusion of iron deposition in the myocardium or other organs in these individuals.
HEREDITARY HAEMOCHROMATOSIS MUTATIONS AND CLINICAL OUTCOME IN DILATED CARDIOMYOPATHY
Analysis of clinical outcomes in relation to the presence of mutations yielded an unexpected finding. Although we did not observe an effect of the H63D allele on the clinical course of disease, carriers of the C282Y allele had milder disease with significantly less cardiac enlargement. Furthermore, there was a trend towards improved survival in patients carrying the C282Y mutation. The significance of this finding is not clear. As these patients were not younger, they would not be expected to have earlier disease than the other groups. It is possible that the C282Y allele is protective by mechanisms as yet unidentified, or it is equally possible that it predisposes to the development of dilated cardiomyopathy in individuals who would otherwise have been disease free. However, the above findings need to be interpreted with caution and require validation in future studies. The observation of milder disease may be a chance finding related to the multiplicity of clinical variables measured, and the study was underpowered to detect a survival advantage of the magnitude observed here. The latter would require 1000 patient years of follow up at five years as opposed to the 259 patient years of follow up in this study.
LIMITATIONS
This study was powered only to determine differences between cases and controls in the frequency of HFE mutations and not to explore mechanisms of association or to determine influences on survival. The hypothesis that the mechanisms of association may relate to factors other than iron is based primarily on available data on the relative effects of H63D and C282Y on iron metabolism,18 with which our own observations are consistent. Unequivocally establishing or excluding that the effect of the variants on the risk of dilated cardiomyopathy is through altered iron metabolism will require a study of much larger size than we have undertaken.
Also, as noted above, this study is underpowered to exclude the possibility that these variants influence outcomes or survival. Furthermore, examination of the influence of HFE mutations on clinical outcomes in patients with dilated cardiomyopathy is limited by the likely heterogeneity of the group. Even within cases classified as familial there is likely to be heterogeneity, depending on the particular genetic mutation responsible for the disease. Unless the underlying groups are similar in other respects, the modulating influence of particular polymorphisms or mutations may be difficult to determine. The use of blood bank controls might also be considered a limitation of this study. These are distinct from the general population in that they are initially self selected volunteers and subsequently undergo further selection by blood bank health screening procedures. Nonetheless the frequency of mutations observed in the controls in this study is consistent with results of large population based studies. Only white donors were used and all analyses were corrected for age and sex differences.
CONCLUSION
We have shown that the frequency of HFE H63D is increased in patients with dilated cardiomyopathy. As H63D has a relatively minor effect on iron status, the mechanism of this association may be unrelated to iron metabolism. The possibility that the association is related to immunological mechanisms should be explored. In addition, further larger studies with clearly defined subgroups are required to determine the modulating influence of HFE mutations in dilated cardiomyopathy.
References
Commentary
Most patients with hereditary haemochromatosis have genetic mutations that determine either a cysteine to tyrosine substitution at position 282 of an HLA-related protein (Cys282Tyr or C282Y) or a histidine to aspartate substitution at position 63 (His63Asp or H63D). Carriers of the “haemocromatosis gene” (HFE) mutation C282Y are reported to be at increased risk of myocardial infarction (Circulation1999;100:1274), speculatively through a mechanism related to the effects of iron handling on free radical generation and atherosclerosis. Heart failure may be a clinical feature of hereditary haemochromatosis. Therefore, this study by Mahon and colleagues explores for the first time the incidences of the C282Y and H63D HFE mutations in patients with idiopathic dilated cardiomyopathy. The results suggest that the frequency of the H63D mutation may be increased in patients with dilated cardiomyopathy. Although the study is not powered to explore mechanistic relations between this mutation and the disease state, it appears at this stage that the effect of the gene mutation is probably manifested independently of effects on systemic iron handling.
Footnotes
↵* N G Mahon and A S Coonar contibuted equally to this work