Article Text

Abstract
Objective: To investigate how permanent inhibition of guanylyl cyclase A receptor (GC-A) affects cardiac function.
Methods: Hearts of GC-A−/− and corresponding wild type mice (GC-A+/+) were characterised by histological, western blotting, and northern blotting analyses. Cardiac function was evaluated in isolated, working heart preparations.
Results: At 4 months of age, GC-A−/− mice had global cardiac hypertrophy (about a 40% increase in cardiac weight) without interstitial fibrosis. Examination of heart function found a significant delay in the time of relaxation; all other parameters of cardiac contractility were similar to those in wild type mice. At 12 months, the hypertrophic changes were much more severe (about a 61% increase in cardiac weight), together with a shift in cardiac gene expression (enhanced concentrations of atrial natriuretic peptide (3.8-fold), B type natriuretic peptide (2-fold), β myosin heavy chain (1.6-fold) and α skeletal actin (1.7-fold) mRNA), increased expression of cytoskeletal tubulin and desmin (by 29.6% and 25.6%, respectively), and pronounced interstitial fibrosis. These changes were associated with significantly impaired cardiac contractility (+dP/dt decreased by about 10%) and relaxation (−dP/dt decreased by 21%), as well as depressed contractile responses to pressure load (all p < 0.05).
Conclusions: Chronic hypertension in GC-A−/− mice is associated with progressive cardiac changes—namely, initially compensated cardiomyocyte hypertrophy, which is complicated by interstitial fibrosis and impaired cardiac contractility at later stages.
- atrial natriuretic peptide
- hypertension
- cardiac hypertrophy
- fibrosis
- cardiac function
- ANP, atrial natriuretic peptide
- BNP, B type natriuretic peptide
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- GC-A, guanylyl cyclase A receptor
- MHC, myosin heavy chain
Statistics from Altmetric.com
- ANP, atrial natriuretic peptide
- BNP, B type natriuretic peptide
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- GC-A, guanylyl cyclase A receptor
- MHC, myosin heavy chain
A rterial hypertension is a major risk factor for cardiovas cular disease. Among other complications the develop ment of left ventricular hypertrophy constitutes an independent risk factor for heart failure and cardiac death. The genetic variations contributing to essential hypertension are largely unknown. Moreover, effective treatment of the increased blood pressure and prevention of secondary morbidity may depend specifically on the underlying genotype, which is probably completely different from patient to patient.
The heart is involved in the endocrine regulation of blood pressure by secreting two natriuretic peptides, atrial (ANP) and B type natriuretic peptide (BNP).1 They activate a common receptor, guanylyl cyclase A (GC-A), which is expressed mainly in the vasculature and kidneys. Subsequent increases in intracellular cyclic GMP initiate vasodilatation, natriuresis, and diuresis, thereby lowering blood pressure.1 Recent studies suggested that these peptides not only maintain blood pressure and volume homeostasis but also are involved locally in moderating the cardiac growth response to hypertrophic stimuli.2 In particular, mice lacking the ANP receptor, GC-A−/−, have pronounced hypertension and cardiac hypertrophy that is disproportionate and partly independent of their increased blood pressure.3–6 These experimental observations emphasise the importance of this system in cardiovascular homeostasis and suggest that some forms of hypertension and cardiac hypertrophy in humans can be explained in part by decreased expression or responsiveness of GC-A. Indeed, blunted vascular and renal effects of exogenous ANP have been shown in patients with cardiac failure or arterial hypertension.7,8 Intriguingly, a recent study showed for the first time the association between a functional deletion mutation in the promoter region of the human GC-A gene and decreased GC-A expression, with essential hypertension and ventricular hypertrophy.9
GC-A−/− mice constitute a powerful experimental model to dissect the cardiovascular functions of the ANP/GC-A system and the consequences of its long term inhibition.3,4 However, until now few data have been available on the impact of GC-A inactivation on cardiac function. To address this issue, in the present study the cardiac performance of GC-A−/− mice at various ages was evaluated in vitro in an isolated working mouse heart preparation. This allowed us to study the effects of GC-A deletion on left ventricular mechanics within a “whole heart” preparation that is independent of systemic compensatory mechanisms. In addition, hearts of GC-A−/− and wild type mice were compared by histological, western blot, and northern blot analyses. Our study shows that genetic deletion of the ANP receptor in mice leads to progressive cardiac changes, with an initially compensated cardiomyocyte hypertrophy associated with interstitial fibrosis and depressed cardiac contractile parameters at later stages. This cardiac phenotype is accompanied by the induction of a fetal gene programme and cytoskeletal alterations in cardiomyocytes that resemble some of the characteristic features of cardiac hypertrophy after long lasting hypertension in humans.
METHODS
Materials
Primary antibodies used for immunohistochemistry and western blot analyses were against myomesin,10 desmin, tubulin, and laminin (Sigma, Buchs, Switzerland), and non-muscle myosin IIB (Chemicon, Temecula, California, USA).11 The secondary antibodies were from Milan (La Roche, Switzerland) and DAKO (Zug, Switzerland). [α-32P]-dCTP and [γ-32P]-ATP were from NEN Life Science Products (Köln, Germany).
Animals
GC-A−/− mice were provided by Dr D L Garbers (HHMI, University of Texas Southwestern Medical Center, Dallas, Texas, USA).3 Male GC-A−/− mice and their non-transgenic (GC-A+/+) littermates, 4 and 12 months old, were used. Genotypes were identified by polymerase chain reaction. Blood pressure was measured in conscious mice by tail cuff (Softron, Tokyo, Japan).12 All investigations conform with the Guide for the care and use of laboratory animals (National Institutes of Health, Publication No 85–23, revised 1996) and were approved by the local animal care committee.
Histology
Hearts were fixed in 4% formaldehyde and embedded in paraffin, and 5 μm sections were stained with periodic acid Schiff (to discriminate cell borders) or 0.1% picrosirius red (for collagen). The mean cardiomyocyte diameter was calculated from photomicrographs of the right and left ventricular free wall by a computer assisted image analysis system (VIDAS 25, Zeiss, Germany) by measuring 100 cells/specimen in the region of the cell nucleus.
Immunohistochemistry and confocal microscopy
Frozen tissue sections (20 μm thick) were cut on a cryostat and collected on gelatin coated slides. Laminin, myomesin, non-muscle myosin heavy chain (MHC) IIB, desmin, and tubulin were stained as previously described.11
Western blot analysis
Frozen ventricles were homogenised and proteins were solubilised in sodium dodecyl sulfate sample buffer and separated on 8–22% gradient polyacrylamide minislab gels (Biorad, Glattbrugg, Switzerland).13 The primary antibodies were against tubulin and desmin.11 The secondary antibodies were horseradish peroxidase conjugated antimouse immunoglobulins. The immunoreactive proteins were visualised by chemiluminescence reaction.11
Northern blot analysis
Messenger RNA concentrations of ANP, BNP, α- and β-MHC, α skeletal actin, and α1(I) procollagen were estimated from 20 μg total RNA. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the sarcoplasmic reticulum Ca2+ storage protein calsequestrin (as myocyte specific gene) were used for normalisation. Specific mouse cDNA probes were labelled with [α-32P]-dCTP; oligonucleotides for β-MHC were labelled with [γ-32P]-ATP. Radioactive signals were visualised in a PhosphorImager and quantified by the ImageQuant software version (Molecular Dynamics, Krefeld, Germany).
Analysis of cardiac function
Mice were anaesthetised intraperitoneally with inactin (10 mg/kg body weight) and the hearts excised. The aorta was cannulated for retrograde perfusion (Langendorff mode) with heated (37.4°C) and oxygenated Krebs-Henseleit buffer, containing (in mmol/l) NaCl (118), KCl (4.7), CaCl2 (2.5), MgSO4, (1.2), KH2PO4 (1.2), sodium EDTA (0.5), NaHCO3 (25), and glucose (11). The pulmonary vein was dissected and a bevelled cannula (PE-50) was passed into the left ventricle and pulled through the ventricular wall. It was anchored in the apex by a fluted end and connected to a pressure transducer. The left atrium was then cannulated through the same pulmonary vein and perfusion of the heart was switched from retrograde to anterograde, fluid ejecting mode.14 Fluid was ejected from the aortic cannula against a hydrostatic fluid column set at a height to yield a mean aortic pressure (afterload) of 50 mm Hg. Atrial inflow (preload) was adjusted to 5 ml/min. Coronary flow (as the difference between preload and aortic flow), heart rate, aortic pressure, and left intraventricular pressure were continuously monitored and the first derivatives of left intraventricular pressure, +dP/dt and −dP/dt (in mm Hg/s), time to peak pressure (ms/mm Hg), and time to half relaxation (ms/mm Hg) were calculated (A Mon 2.1 program, Ingenieurbüro Jäckel, Hanau, Germany).14
Statistical analysis
Results are expressed as the mean (SEM). Differences between GC-A−/− and +/+ mice were determined with an unpaired Student's t test. Probability values of p < 0.05 were considered significant.
RESULTS
Hypertension and cardiac enlargement
In agreement with previous studies, homozygous GC-A−/− mice had an average increase in systolic and diastolic blood pressures of 28 (1.5) and 16 (2) mm Hg, respectively (n = 23 for each genotype, p < 0.01).3,4,12 We found no significant differences between blood pressures dependent on age (earliest recordings at 6 weeks, latest at 13 months of age) or sex. The heart to body weight ratios of GC-A−/− mice were significantly increased, averaging 140 (2.1)% of wild types by 4 months and up to 162 (1.7)% of wild types by 12 months of age. Both the left ventricular to body weight ratios (126 (2.8)% and 136 (3.2)% of GC-A+/+ at 4 and 12 months of age, respectively) and the right ventriclat to body weight ratios (148 (2.6)% and 159 (3.8)%) were significantly increased (n = 23 for each genotype, p < 0.01).
Cardiomyocyte hypertrophy
Morphometrical analyses of hearts from 12 month old mice showed severe cardiomyocyte hypertrophy in both GC-A−/− ventricles, with the following cellular diameters: left ventricle, 15.1 (0.8) μm (−/−) v 12.1 (0.3) μm (+/+); right ventricle, 13.4 (0.6) μm (−/−) v 10.4 (1.2) μm (+/+) (n =6; p < 0.05). To elucidate whether this increase in cardiomyocyte width is progressive, ventricular sections from mice aged 4 and 12 months were analysed by confocal microscopy (n = 4) (fig 1A–D). The lateral borders of the cardiomyocytes were visualised by staining of the extracellular matrix component laminin; the cardiomyocytes themselves were stained with an antibody to sarcomeric myomesin. Significant increases in cardiomyocyte width were observed in 4 month old and 12 month old GC-A−/− mice, and were more pronounced in the latter (fig 1B, D). The ultrastructure of the cardiomyocytes as visualised with myomesin seemed to have been unaffected.

Confocal micrographs taken from guanylyl cyclase A receptor (GC-A) +/+ (A, C, E, G) and GC-A−/− hearts (B, D, F, H) at 4 months (A, B, E, F) and 12 months (C, D, G, H) of age (n = 4). Sections A to D were double stained with antibodies to laminin (green, to visualise the cell borders) and myomesin (red, to show cross striations in the cardiomyocytes). Sections E to H were stained with an antibody to non-muscle myosin heavy chain IIB (green, to visualise cardiac fibroblasts). A progressive increase in cardiomyocyte width can be observed in the GC-A−/− hearts (compare arrows in B and D). This was accompanied by an increased number of fibroblasts at 12 months of age (H). Bars represent 10 μm (A to D) and 20 μm (E to H).
In addition to cellular hypertrophy, an increase in the cytoskeletal proteins of the microtubules and the intermediate filaments was found (fig 2). In hearts from 12 month old GC-A−/− mice, western blot analyses showed an increase of tubulin by 29.6 (1.5)% and of desmin by 25.6 (2)% (n = 5, p < 0.05) (fig 2B). Immunohistological analysis of frozen sections showed that the increase of desmin was clearly more pronounced at the intercalated disks, whereas the increase in tubulin was more generalised (fig 2A).

Analysis of cytoskeletal proteins by immunohistochemistry (A) and western blotting (B). (A) Confocal micrographs of left ventricles from 12 month old GC-A+/+ (a, c) and GC-A−/− mice (b, d). Sections were double stained with antibodies to desmin (a, b) and tubulin (c, d). For desmin, similar striations at the Z disks can be observed in the GC-A+/+ and −/− hearts, but the signal is increased at the intercalated disks of GC-A−/− hearts (arrows). For tubulin, a general upregulation of signal was observed in the GC-A−/− hearts. (B) Western blotting showing increased expression of desmin and tubulin in GC-A−/− ventricles compared with GC-A+/+ (n = 5). *p < 0.05. Bars represent 20 μm.
Cardiac fibrosis
In hearts from 4 month old GC-A−/− mice, northern blot analysis showed a slight increase in α1(I) procollagen mRNA by 143 (4)%, suggesting increased activity of fibroblasts (n = 12, p < 0.05) (fig 3B). Nevertheless, this was not associated with obvious interstitial fibrosis, as shown by sirius red staining of paraffin sections (n = 5) (fig 3A). In contrast, at 12 months of age the GC-A−/− hearts showed focal fibrosis mostly in subendocardial regions (n = 5) (fig 3A). This was corroborated by northern blot analyses, showing a 4.2-fold increase in α1(I) procollagen mRNA (n = 12) (fig 3B). The increased number of fibroblasts in the hearts of 12 month old GC-A−/− mice was also visualised with confocal microscopy by staining for non-muscle MHC IIB, a myosin isoform that is rarely expressed in cardiomyocytes (n = 4) (fig 1E–H).

Cardiac fibrosis in hearts of 4 and 12 month old GC-A−/− mice was estimated by histology using picrosirius red staining (A) and by northern blot analysis of α1(I) procollagen mRNA expression (B). (A) Representative sections showing focal fibrosis in hypertrophied left ventricles obtained from 12 month old GC-A−/− mice; these changes were not observed in 4 month old GC-A−/− mice or in age matched wild type (GC-A+/+) controls (n = 5). Bars represent 50 μm. (B) The α1(I) procollagen transcripts were normalised to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (n = 12). *p < 0.05.
Cardiac gene expression
Cardiac hypertrophy in GC-A−/− mice was associated with increased mRNA expression of the following markers in 4 month old and 12 month old GC-A−/− hearts, respectively: ANP, by 2.3 (0.4)*-fold and 3.8 (0.5)*-fold; BNP, by 1.8 (0.2)*-fold and 2 (0.2)*-fold; β-MHC by 1.3 (0.2)-fold and 1.6 (0.2)*-fold; and α skeletal actin by 1.3 (0.1)-fold and 1.7 (0.1)*-fold (n = 6 to 12, *p < 0.05 v age matched GC-A+/+; fig 4).

(A) Representative northern blot analyses showing the expression of atrial (ANP) and B type natriuretic peptide (BNP), α skeletal actin, β (β-MHC) and α myosin heavy chain, and (for reference) calsequestrin (CSQ) and GAPDH in the hearts of 12 month old GC-A+/+ and −/− mice. (B) GC-A−/− hearts show increased mRNA expression of ANP, BNP, α skeletal actin (α-sk), and β-MHC. The transcripts were normalised to the expression of CSQ, as cardiomyocyte specific gene, and compared with the expression level in GC-A+/+ mice (n = 12). *p < 0.05.
Cardiac function
To evaluate cardiac function, we used the isolated working heart preparation. Initially, the GC-A−/− and corresponding GC-A+/+ hearts were compared under identical afterload (50 mm Hg) and preload conditions (5 ml/min). In hearts from 4 month old GC-A−/− mice the time of relaxation was prolonged by 12 (2)% (n = 5, p < 0.05); all other cardiac contractile parameters were not significantly different from GC-A+/+ (table 1). In the hearts from 12 month old GC-A−/− mice both contractility and relaxation were impaired (table 1). Left ventricular systolic pressure was reduced by 7%, +dP/dt was decreased by 10%, and −dP/dt was decreased by 21% compared with GC-A+/+ hearts (all p < 0.05) (table 1). The time to peak pressure was unchanged, while the time of relaxation and time to half relaxation (ms/mm Hg) were prolonged by 18% and 28%, respectively (both p < 0.05; table 1). Thus, particularly the diastolic function of the GC-A−/− hearts was significantly impaired.
Contractile parameters of isolated hearts from 4 and 12 month old guanylyl cyclase A receptor (GC-A) +/+ and −/− mice in the working heart preparation.
Effect of pressure loading
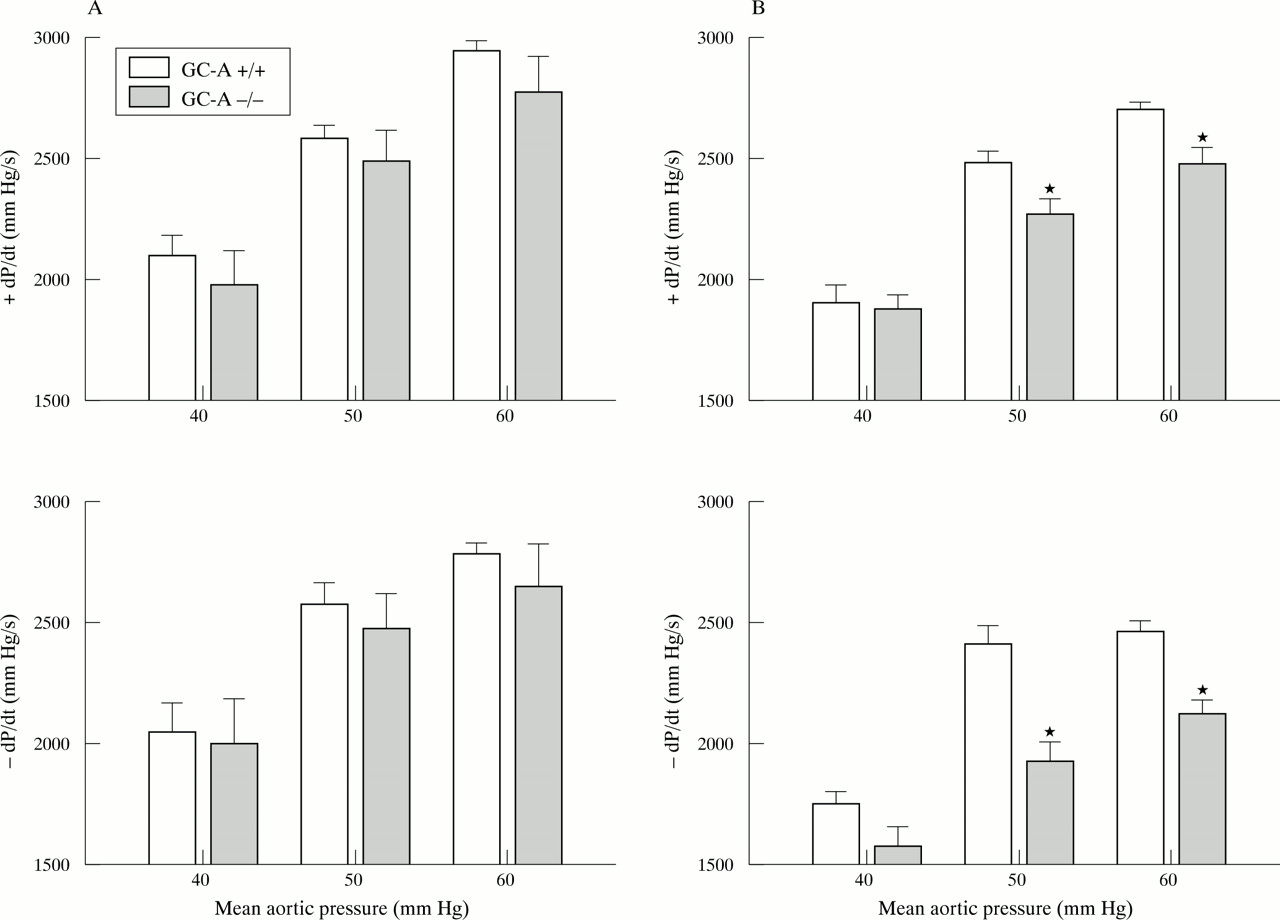
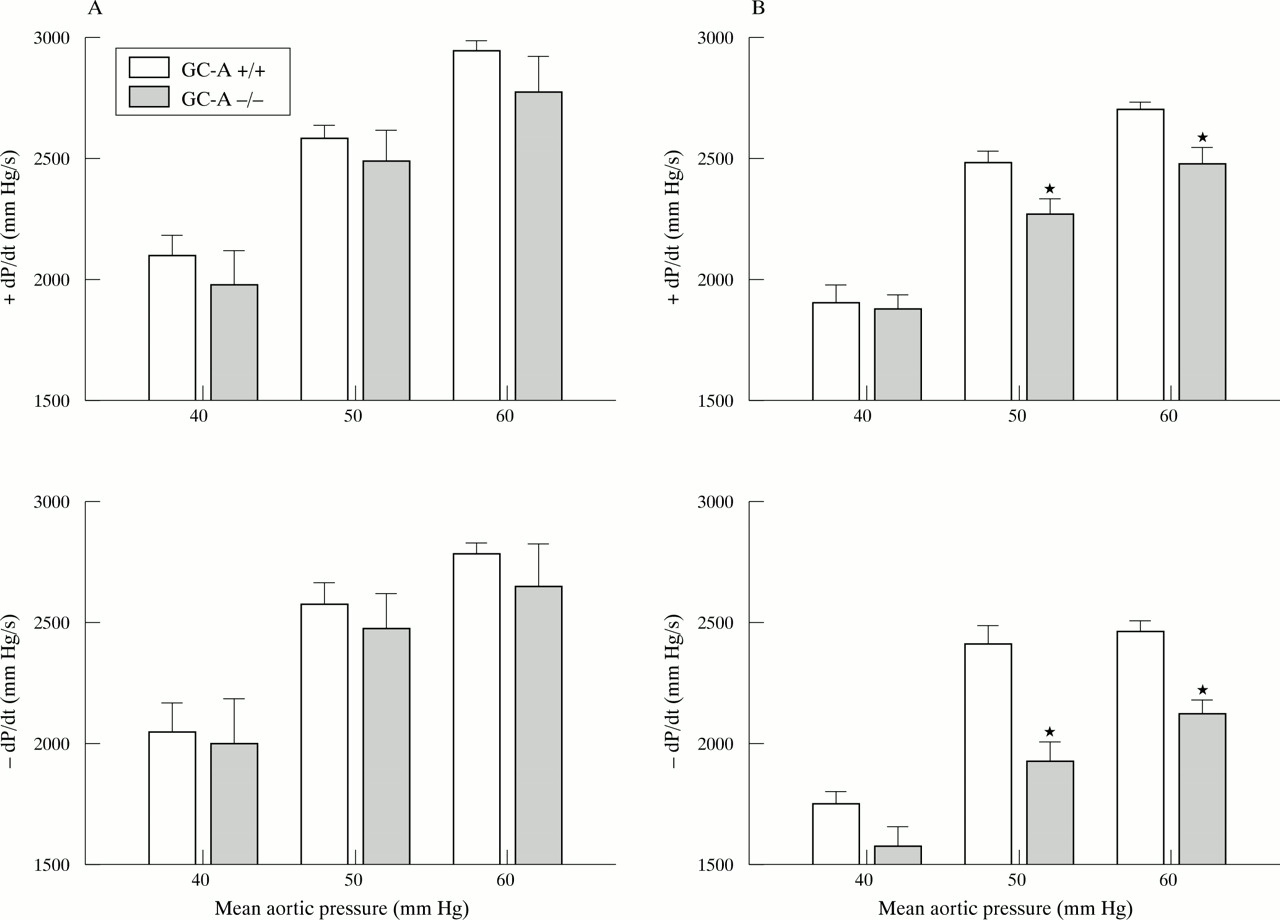
The left ventricular contractile responses to increased aortic pressure were determined by changing the height of the afterload fluid column. At 4 months of age the contractile responses of GC-A−/− hearts and GC-A+/+ hearts were comparable (n = 5 for each genotype). At 12 months of age, the contractile function of GC-A−/− hearts was greatly reduced, with significantly lower maximal rates of contraction and relaxation for all pressure loading conditions (n = 8 for each genotype) (fig 5). Also, at this later age the increases in left ventricular systolic pressure in response to increased pressure load were significantly decreased in the GC-A−/− hearts (GC-A+/+ v GC-A−/−): 70.7 (1) v 69 (1.8) mm Hg (afterload 40 mm Hg), 88 (1.2) v 82 (2.1)* mm Hg (afterload 50 mm), 96 (0.9) v 90 (2.1)* mm Hg (afterload 60 mm) (*p < 0.05 v GC-A+/+).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Isolated working heart preparations showing the effect of pressure loading on the contractile function of GC-A+/+ and −/− hearts obtained from (A) 4 month old (n = 5) and (B) 12 month old mice (n = 8). Hearts were perfused at 5 ml/min (preload). Aortic pressure (afterload) was sequentially adjusted by changing the height of the afterload fluid column. Left ventricular function is plotted as afterload (mm Hg) versus the rate of contraction (+dP/dt in mm Hg/s, top) and the rate of relaxation (−dP/dt in mm Hg/s, bottom). *p < 0.05 v GC-A+/+.
DISCUSSION
Our results show that chronic hypertension in GC-A deficient mice is associated with severe cardiac changes—namely, progressive cardiomyocyte hypertrophy, alterations in the expression of sarcomeric and cytoskeletal proteins, a shift in cardiac gene expression, and at a later stage interstitial fibrosis. These changes ultimately lead to alterations in basal cardiac contractile parameters and decreased contractile responsiveness to cardiac pressure load, as assessed in isolated working hearts.
Cardiac hypertrophy and fibrosis
The following suggests that cardiomyocyte hypertrophy in GC-A−/− mice is not simply an adaptive response to chronic hypertension and that other stimuli contribute to myocyte growth in this genetic model. Integration of data obtained by confocal microscopy showed that GC-A−/− mice already have increased cardiomyocyte volumes at birth (in GC-A+/+ v GC-A−/− mice, respectively: left ventricle, 1248 (92) μm3v 1648 (111) μm3; right ventricle, 1128 (69) μm3v 1506 (127) μm3; n = 6, p < 0.05). In addition, other transgenic mice with similar increases in blood pressure do not have this hypertrophic phenotype.15 ANP and BNP inhibit growth and proliferation of cultured cardiac myocytes and fibroblasts through GC-A.2 Moreover, two recent reports showed for the first time that the ANP/GC-A system exerts local antihypertrophic effects in vivo.5,6 Mice lacking BNP do not display increased blood pressure or cardiac hypertrophy but they develop cardiac fibrosis, suggesting a local antifibrotic role for BNP that is possibly also mediated through GC-A.16 Thus, inhibition of GC-A abolishes the local growth moderating effects of ANP and BNP and thereby accelerates the development of cardiac hypertrophy and fibrosis in response to hypertension. As shown, myocyte hypertrophy is even more pronounced in the right GC-A−/− ventricles, apparently in the absence of pulmonary hypertension.17,18 This again emphasises the important local role of the ANP/GC-A signalling pathway in preventing cardiac hypertrophy.
Cardiac dysfunction
Our main goal was to determine how these changes in GC-A−/− mice affect cardiac function. As shown, the contractile parameters from isolated working hearts of 4 month old GC-A+/+ and −/− mice were not different between genotypes, with the exception of a slightly delayed relaxation time. Indeed, previous studies by echocardiography in anaesthetised GC-A−/− mice showed that at young ages their hearts are fully functional, despite cardiac hypertrophy.4 In contrast, isolated working hearts obtained from 12 month old GC-A−/− mice develop a lower intraventricular peak pressure and have decreased rates of contraction and relaxation and prolonged time to relaxation even under resting conditions. In addition, the contractile responses to pressure loading are greatly reduced, showing a significant systolic and, even more, diastolic dysfunction. Unfortunately, in the aforementioned echocardiographic report, cardiac haemodynamics were not evaluated in older GC-A−/− mice. In a second study done by magnetic resonance imaging, resting left ventricular volumes and ejection fractions were not different between GC-A+/+ and −/− mice.19 However, this study mainly assessed the accuracy of this technique for the evaluation of left ventricular mass; cardiac function was determined only in two animals at each age and statistical evaluation therefore seems difficult.
In the study by Oliver and colleagues4 sudden death, sometimes with morphological evidence of congestive heart failure, occurred in all GC-A−/− males before six months of age. We did not observe this high death rate; only about 15% of the GC-A−/− mice died spontaneously before 12 months of age, compared with about 5% of wild type littermates. The cause of death in GC-A−/− mice was not established, although necropsy found pronounced cardiac hypertrophy and cardiac interstitial fibrosis in each case. We have no explanation for the difference in the death rates observed between the GC-A−/− colonies. It is noteworthy that basal blood pressures of GC-A+/+ and −/− mice were greatly higher in the study by Oliver and colleagues.4 Although both colonies arose from a mixed 129SV × C57bl6 background, significant differences may exist in the genetic background to account at least in part for the observed differences.
Cardiac changes contributing to dysfunction of GC-A−/− hearts
Within the cardiomyocytes, the observed isoform shift of contractile proteins, with increased expression of α skeletal actin and β-MHC, as well as the increase in the cytoskeletal proteins tubulin and desmin, may be an attempt to compensate cardiac function in the presence of chronic pressure overload.20,21 Accumulation of tubulin and desmin has also been described in human failing hearts, probably to counteract the increased strain on the myocardium.22 This in turn may lead to cytoskeletal stiffness and contractile dysfunction, in particular impaired diastolic relaxation, before the transition to overt heart failure occurs.22 In addition, increased interstitial fibrosis very likely contributes to cardiac dysfunction in the older GC-A−/− mice. Remodelling of the collagen network may affect diastolic myocardial function even more than myocyte hypertrophy.23
Advantages and limitations of the study
The requirement of anaesthesia limits the evaluation of cardiac function in many animal models; peripheral vasodilatation, effects on central sympathetic output, and the direct chronotropic effects of some anaesthetics may all modify ventricular function. In addition, specific neuroendocrine parameters known to be modulated by the ANP/GC-A system, such as sympathetic tone, may be altered in GC-A−/− mice and in turn affect cardiac function in vivo.24 Our primary focus was to assess the intrinsic ventricular mechanics of GC-A−/− hearts. The isolated working heart preparation offers a controlled picture of left ventricular function and of the contractile responses to changes in pressure load. A major limitation is the impossibility of assessing right ventricular dynamics, which will be an issue for our future studies.
Summary
In humans, loss of function of the ANP receptor, GC-A, has been described in at least two different clinical situations. In patients with heart hypertrophy or heart failure the plasma concentrations of ANP are increased but the systemic responses to the peptide are blunted, suggesting decreased responsiveness of GC-A.7 Moreover, a recent study showed that a functional deletion mutation in the human GC-A gene and decreased receptor activity are associated with essential hypertension and ventricular hypertrophy.9 In view of the results of the present study clearly showing the severe cardiac consequences of GC-A dysfunction, the relevance of decreases in the function of ANP or GC-A specifically to human patients must await further work.
Acknowledgments
The authors thank Astrid Grevelhörster and Stephan Lange for excellent technical assistance. This study was supported by the German Bundesministerium fuer Bildung und Forschung (grant BMBF 01EC9801 to MK), the Deutsche Forschungsgemeinschaft (SFB 556 to MK), and the Swiss National Science Foundation (grant 31.52417/97 to JCP).