Article Text

Abstract
Objective: To assess the clinical validity of polymerase chain reaction (PCR) based molecular methods in the microbiological diagnosis of culture negative infective endocarditis in a group of surgically treated patients.
Design: Retrospective case–control study.
Setting: Reference cardiovascular surgical centre.
Patients and samples: 15 culture negative patients with infective endocarditis classified according to Duke criteria, with 17 heart valve samples; 13 age and sex matched control patients without infective endocarditis, with 13 valve samples.
Interventions: Medical records were reviewed and clinical, demographic, and microbiological data collected, including results of molecular detection of bacteria and fungi from valve samples. The clinical validity of molecular diagnosis was assessed, along with the sensitivity and speed of the systems.
Results: In the study group, 14 patients were PCR positive (93%). Organisms detected were streptococci (3), staphylococci (2), enterobacter (1), Tropheryma whippelii (1), Borrelia burgdorferi (1), Candida albicans (1), and Aspergillus species (2). Three cases were positive on universal bacterial detection but the pathogen could not be identified because of contaminating background. One case was negative. All but two positive cases showed clinical correlations. These two cases had no symptoms of infective endocarditis but there was agreement with the surgical findings. All control cases were PCR negative. Results were available within eight hours, and if sequencing was necessary, within 48 hours.
Conclusions: PCR based molecular detection of pathogens in valve samples from surgically treated culture negative infective endocarditis patients is fast, sensitive, and reliable. The technology, combined with thorough validation and clinical interpretation, may be a promising tool for routine testing of infective endocarditis.
- infective endocarditis
- molecular biology
- polymerase chain reaction
- AFLP, amplified fragment length polymorphism
- PCR, polymerase chain reaction
- PVE, prosthetic valve endocarditis
- RFLP, restriction fragment length polymorphism
- UNB, universal bacterial detection
- UNF, universal fungal detection
Statistics from Altmetric.com
- AFLP, amplified fragment length polymorphism
- PCR, polymerase chain reaction
- PVE, prosthetic valve endocarditis
- RFLP, restriction fragment length polymorphism
- UNB, universal bacterial detection
- UNF, universal fungal detection
Targeted antibiotic treatment is the ideal approach to the pharmacological management of infective endocarditis, so the identity of the pathogen causing the disease must be determined whenever possible.1 Patients with culture negative infective endocarditis have a greater frequency of complications than culture positive patients, though mortality seems to be similar.2
Factors that influence the rate of culture negativity in infective endocarditis include failure to use appropriate culture technology, the use of antibiotics before the collection of blood culture samples, and infections caused by fastidious or non-culturable pathogens.3–5
Among culture negative cases of infective endocarditis, one particularly important population group is patients undergoing surgical treatment. These patients have in most cases been treated with antibiotics (and many have received several courses), have prosthetic valve infection, are graft carriers or transplant patients, or have developed rapidly progressive infective endocarditis.2,6 Thus it is not surprising to find a high rate of culture negativity in this group. On this basis, an accurate and rapid aetiological diagnosis, along with timely surgical treatment if indicated, is of the greatest importance in such patients.
New culture methods and improved media have been developed to aid the detection of previously non-culturable or fastidious microorganisms causing infective endocarditis.3 Nevertheless, many cases remain culture negative and these present a challenge to physicians because of their high morbidity and mortality.2
Several molecular approaches have been assessed for detecting and identifying pathogens in a wide variety of infectious diseases. Among these, the polymerase chain reaction (PCR) has perhaps been the most widely used method in recent years and is a powerful aid to microbiological diagnosis. Various investigators have explored the ability of PCR to detect the nucleic acids of fastidious and non-culturable agents in blood and heart valves from patients with infective endocarditis,7,8 and it has been shown to be both robust and accurate in the identification of specific pathogens.3 The possibility of including molecular diagnosis related criteria as part of the currently used Duke scheme for the diagnosis of infective endocarditis has also been suggested.9
However, with the exception of a few PCR specific assays, the molecular diagnosis of infective endocarditis remains a research tool. Aspects such as the specificity and reproducibility of results, the quantitative precision of the measurements, and the financial implications have prevented this technology from coming into regular use in the clinical setting.4 In this paper, we assess the clinical applicability of PCR based methods in the detection of the causative pathogens involved in infective endocarditis in a strictly defined group of culture negative, surgically treated patients.
METHODS
Study design
This was a retrospective case–control study. Medical records from all patients admitted to our institution with a diagnosis of infective endocarditis during the years 1999 to 2001 were reviewed, and patients who had both negative blood cultures and surgical treatment were included in the study. Demographic, clinical, and microbiological data, together with results of molecular diagnosis, were extracted to form a tailored database for further analysis. Molecular diagnosis data were also collected from an age and sex matched control group comprising patients who underwent valve replacement for reasons other than suspected infective endocarditis.
Patients and samples
Study group
Seventeen valve samples were procured during valve replacement, corresponding to 15 culture negative, surgically treated patients with infective endocarditis. Infective endocarditis was defined according to Duke’s clinical criteria.10 Culture negativity was defined as no growth of microorganisms from blood cultures or growth of pathogens suggestive of contamination in fewer than two blood cultures.10,11
Control group
Thirteen valve samples were obtained from 13 patients who were undergoing valve replacement for reasons other than suspected infective endocarditis. Control cases were selected to match the study group for sex and age.
Molecular diagnosis
Sample acquisition
At the time of valve replacement, the infected valve was removed, with strict observance of aseptic conditions. Two experienced surgeons resected representative pieces of valve or the entire diseased native or prosthetic valve. Decisions about the tissue to be resected were left to the surgeons, although vegetations and abscesses were always resected and sent for molecular testing. The valves or portions of them were collected in sterile bottles without fixatives and delivered for molecular detection of pathogens.
DNA isolation
A piece of the valve, vegetation, or abscess of approximately 4 × 4 mm (in suspected native valve infection) was cut into small pieces using a sterile scalpel. This was followed by digestion with a lytic enzyme (lysozyme at a final concentration of 0.4 mg/ml (Roche Diagnostics, Mannheim, Germany) for one hour at 37°C and protease treatment at 56°C for 30 minutes (proteinase K, final concentration 1 mg/ml ; Carl Roth, Germany). A rapid and, in our experience, consistently efficient silica based DNA isolation process (DNA isolation kit, Genotyp s.r.o., Brno, Czech Republic) was immediately performed.
Prosthetic valves were carefully scraped with a scalpel and then vigorously washed in sterile physiological solution. The suspension obtained was enzymatically digested and DNA isolated as above.
PCR
For universal bacterial detection (UNB) we used two primers targeting conserved sequences at the 3′ end of the 16S rRNA bacterial gene (forward primer RW01: 5′ aac tgg agg aag gtg ggg at 3′; reverse primer DG74: 5′ tgc ggt tgg atc acc tcc t 3′). The 50 μl PCR reactions contained the following ingredients: 1 × HotStart Taq Master Mix (Qiagen, Hilden, Germany), 12 μl of distilled water, 15 pmol of each primer, 0.7 mM MgCl2, 100 mM of dUTP (Roche Diagnostics, Mannheim, Germany), and 8-methoxypsoralen at a final concentration of 25 μg/ml. After incubation at 4°C for 30 minutes, the vials were decontaminated by ultraviolet irradiation at 4 J/cm2 for four minutes. Finally, 3 μl of DNA template were added into the decontaminated PCR vials and the PCR performed in a PTC-200 thermal cycler (MJ Research, Watertown, Massachusetts, USA).
Cycling for UNB detection was set as follows: an initial cycle at 94°C for 15 minutes for polymerase activation, then 45 cycles consisting of a denaturation step at 96°C for 10 seconds, an annealing step at 58°C for 10 seconds, and a synthesis step at 72°C for 30 seconds. A last extension step at 72°C for 10 minutes was included after the 45 amplification cycles. The amplified 370 base pair (bp) product is variable enough to allow discrimination by restriction fragment length polymorphism analysis (RFLP) of the most prevalent causative agents of infective endocarditis.
For universal fungal detection (UNF), an equimolar mixture of forward primers was used (UNF1, 5′ gca tcg atg aag aac gca gc 3′; UNF1A, 5′ gca acg atg aag aac gca gc 3′; UNF1B, 5′ gca tcg atg aag aac gta gc 3′) along with the reverse primer UNF2 (5′ ttg ata tgc tta agt tca gcg g 3′). These primers target conserved sequences at the 3′ end of the 5.8S rRNA gene and the 5′ end of the 25S subunit of the fungal ribosomal DNA, including a part of the internal transcribed spacer 2 (ITS2) region. The 50 μl PCR mixture contained 1 × HotStart Taq Master Mix (Qiagen), 15 pmol of the mixture of forward primers, 15 pmol of UNF2 primer, 100 μM of dUTP (Roche Diagnostics), and 5 μl of template DNA.
PCR cycling was as follows: (1) incubation at 96°C for 12 minutes for activation of DNA polymerase; (2) 40 cycles consisting of a denaturation step of 10 seconds at 96°C, an annealing step at 58°C for 10 seconds, and an extension step at 72°C for 30 seconds; and (3) a final extension step at 72°C for four minutes. The length of the amplified segment varied from 195 to 544 bp and permitted species level discrimination of most medically important fungi after treatment with a set of restriction enzymes.
An internal standard was constructed using PCR MIMICS (competitive PCR fragments)12 and added into each PCR vial. This internal standard, designed for use in both UNB and UNF systems, is amplified as a 519 bp product that permits competitive amplification with the DNA template. Positive controls for Gram positive bacteria (Staphylococcus aureus) and Gram negative bacteria (Escherichia coli), as well as a total negative control (PCR mix only, without template and internal standard), a negative control (with distilled water instead of DNA template), and isolation controls (distilled water instead of tissue sample) were added into our routine UNB detection strips. For UNF testing, positive (Candida albicans) and isolation controls were used. These controls enabled monitoring of PCR performance and specificity, as well as checking for crossover and carryover contamination.
Amplified fragment length polymorphism
Amplified UNB and UNF PCR products were loaded into ethidium bromide stained agarose gel, further separated by electrophoresis, and visualised by ultraviolet transillumination at 312 nm (fig 1). Amplified fragment length polymorphism (AFLP) analysis, in which the length of the PCR fragment is determined with the help of a molecular size marker, was accurate enough to establish the identity of several fungal pathogens. However, as the fragment obtained after PCR amplification in samples positive for bacterial infection has a unique length, AFLP was not useful for UNB identification.

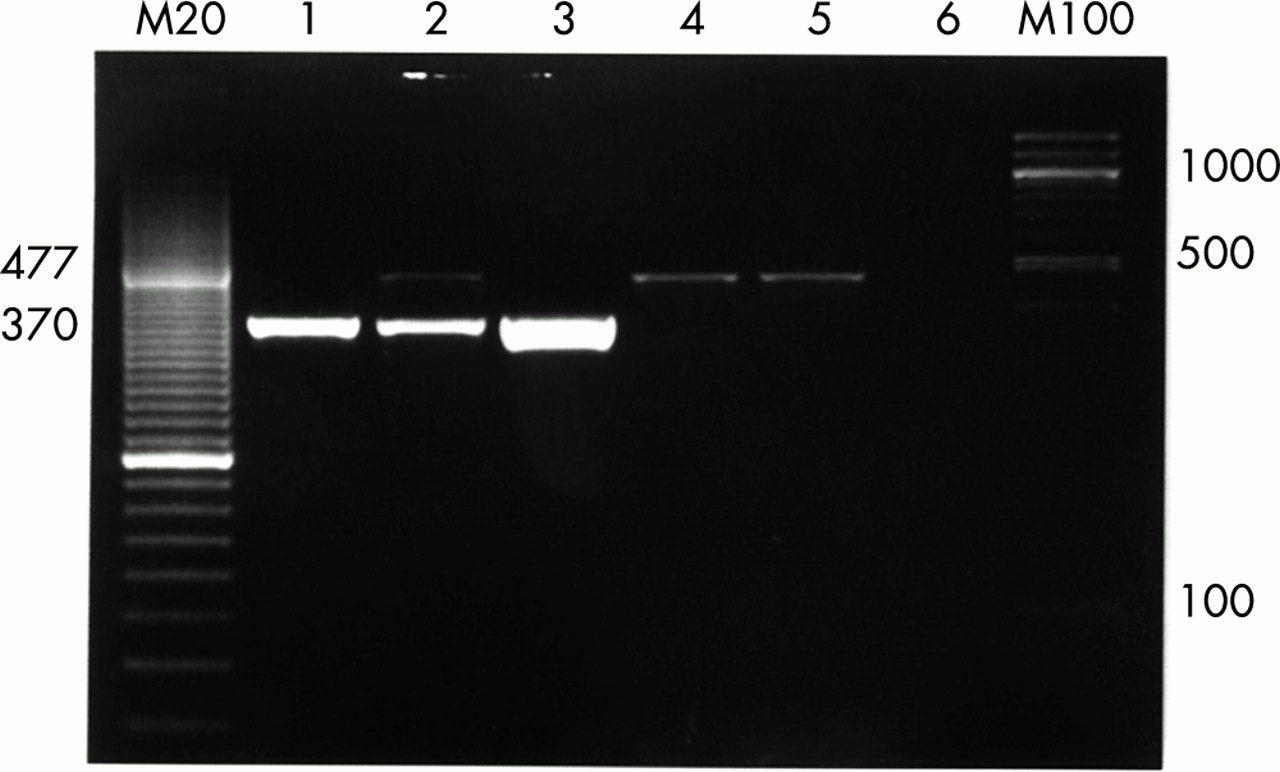
Photograph of gel electrophoresis of UNB products. Lane 1: positive control S aureus, 103 copies/μl; lane 2: positive control E coli, 103 copies/μl; lane 3: positive sample; lane 4: negative control, a band of 477 base pairs (bp) corresponding to internal standard, 103 copies/μl, is shown; lane 5: isolation control, the internal standard 477 bp product appears again; lane 6: total negative control. M20 and M100, molecular size markers (20 bp and 100 bp).
Restriction fragment length polymorphism
Positive samples were digested with restriction endonucleases (Hae III and Nco I for UNB identification; Hae III and Taq I for UNF identification; New England Biolabs, Beverly, Massachusetts, USA) and the resulting fragments separated on gel electrophoresis as described above (fig 2). In several important bacterial and most fungal infective endocarditis pathogens, the fragments obtained correspond to specific electronically predictable patterns (BLAST algorithm, NCBI; WebCutter 2.0, Max Heiman, 1997). Comparison of the patterns obtained with those predicted indicates the identity of the detected pathogens. RFLP may therefore permit full discrimination of most medically important fungi and partial identification (at the group or genus level) of bacterial pathogens causing infective endocarditis.

{kind=link}
{kind=link}
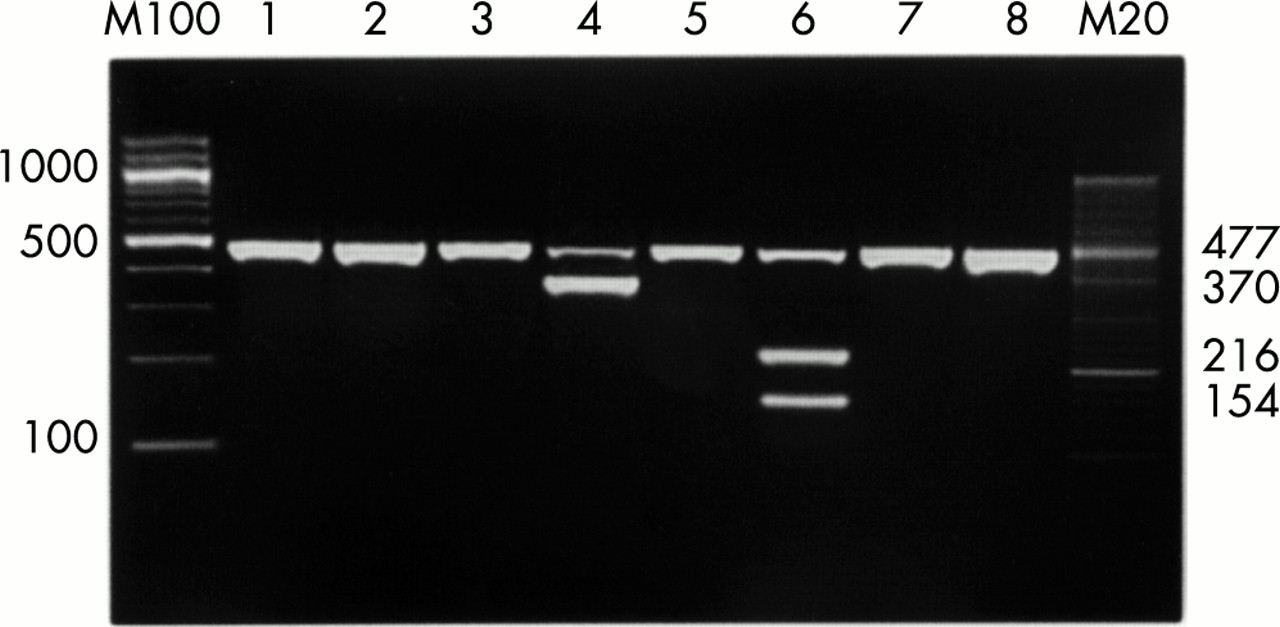
Photograph of gel electrophoresis of UNB-RFLP products after Hae III treatment. Lane 4: an amplification product that corresponds to Staphylococcus species is not digested by Hae III; lane 6: a digestion pattern that corresponds to Streptococus species. M20 and M100, molecular size markers (20 and 100 base pairs).
Specific assays and sequencing
In a few cases, specific assays (S aureus, Staphylococcus epidermidis, Borrelia burgdorferi sensu lato) were used. In unclear cases, however, PCR products were sequenced (Sequencing Services, VBC Genomics, Bioscience Research GmbH, Vienna, Austria) and compared with sequences obtained from public domain databases (Entrez-Nucleotide, NCBI) for determination of pathogen identity.
RESULTS
Case definitions
Of the 15 patients included in the study, 12 were classified as “possible” infective endocarditis and three were “definite” cases. Eleven of the “possible” cases were classified as such because they did not fulfil the major blood culture positivity criterion. The remaining “possible” case showed no major criteria, but had two minor criteria. One “definite” case fulfilled two major criteria (new heart murmur and ultrasound specific findings) and one minor. The remaining two “definite” cases fulfilled one major criterion (cardiac ultrasound specific findings), and three minor criteria in one case and four in the other. No case was reclassified as “rejected” infective endocarditis in the follow up.
Detection of pathogens
All samples were tested with both bacterial and fungal universal detection systems. The results of molecular testing along with the characteristics of the patients studied are shown in table 1. PCR detected DNA from bacteria or fungi in 14 of the 15 suspected cases of infective endocarditis (93%). Eleven cases corresponded to bacteria and three to fungi. Identified bacteria were as follows: Streptococcus species (3), Staphylococcus species (2) (one S aureus and one S epidermidis), Enterobacter species (1), and B burgdorferi sensu lato (1). Tropheryma whippelii was identified in one case by sequencing the amplified product, which became positive on UNB. The remaining three positive cases showed positivity on UNB but owing to contaminating background it was impossible to identify the pathogen by RFLP analysis or to perform further sequencing of the PCR products. These samples were labelled as positive “non-defined” cases.
Characteristics of the patients: clinical features and molecular diagnosis
Among the three cases in which fungi were detected, Candida albicans was identified in one and Aspergillus species in two.
All samples in the control group were PCR negative on UNB and UNF detection.
Clinical validity
Patient 4 was found to have T whippelii endocarditis without gastrointestinal manifestations of Whipple’s disease. Samples from patient 7 were positive on a specific assay for B burgdorferi sensu lato. This patient had strong positivity for borrelia specific antibodies in blood. Three patients showed fungal endocarditis, two of whom (patients 1 and 11) had prosthetic valve infective endocarditis (PVE) episodes and the third (patient 3) was a kidney transplant patient in whom disseminated aspergillosis developed. All PCR results from the remaining patients showed clinical correlations except in two cases who had no clinical manifestations of infection. PCR results demonstrating the presence of pathogens correlated with surgical findings of infection (valve perforation in one case and vegetation in the other) in these latter two cases.
Speed
The detection systems enabled us to process clinical samples and report PCR results within eight hours after sample delivery to the laboratory. DNA isolation took one to two hours, PCR another one and a half to two hours, and RFLP, three more hours, while gel electrophoresis added on average one hour to the procedure. When sequencing of the amplicon was necessary, the results were available within 48 hours.
DISCUSSION
The rate of culture negative infective endocarditis varies among different studies, ranging from 2.5–31% of all cases.4,13–17 Many of these patients will undergo surgery for reasons that have already become established as sound indications for the surgical treatment of infective endocarditis (prosthetic valve endocarditis, antibiotic non-responsiveness, acute infection with rapidly developing heart failure, recurrent embolism, heart block caused by infection, abscess formation).17 Patients with culture negative infective endocarditis have often been pretreated with antibiotics, thus preventing culture identification of the pathogen.
In prosthetic valve endocarditis and in immunocompromised patients, fungal infections account for 2–10% of all cases.18 Additionally, fastidious bacteria and pathogens such as the HACEK group (Haemophilus species, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis, Eikenella corrodens, and Kingella kingae), which are often difficult to culture, are likely causes of infective endocarditis in this group of patients. Thus the probability of identifying those pathogens in blood cultures or in cultures from valves can be low.3,19
Culture independent molecular methods have aided microbiological diagnosis in culture negative infective endocarditis. Amplification of the 16S rRNA bacterial gene has enabled the identification of various pathogens causing infective endocarditis, including species of coxiella, legionella, chlamydia, bartonella, and T whipelii, among others.3,7,20–22 Fungi have also been identified by amplification of targeted sequences in the fungal ribosomal DNA.23,24 This has enabled the diagnosis of infection caused by rarely cultured or non-culturable organisms such as Aspergillus species.25,26
Broad range (universal) detection has been useful in infective endocarditis.8,27 However, few studies have addressed the value of these detection systems directly in clinical settings and on clinical samples.27,28 In 1997 Goldenberger and colleagues applied a universal bacterial detection system to bacterial isolates from clinical samples and showed a good correlation with culture results.8
Apart from its high sensitivity, one of the advantages of PCR is that it is a rapid and reliable form of molecular diagnosis in infective endocarditis. The next step should be to explore these advantages by testing the direct applicability of the method in clinical settings. We have therefore assessed and validated the use of PCR based universal methods for detecting pathogens in a clinical infective endocarditis setting on a defined group of culture negative patients.
The Duke criteria have been shown to be a reliable tool in the diagnosis of infective endocarditis.15,29,30 In our study none of the suspected infective endocarditis cases was reclassified as “rejected” infective endocarditis on the follow up, so the diagnostic validity of the Duke scheme was considered adequate for clinical definition of the patients included in this investigation.
The microbiological spectrum of native valve infective endocarditis cases after molecular testing corresponds to the pathogens that commonly cause infective endocarditis, with the following two exceptions.
T whipelii was identified in patient 4, who had no particular findings in the past history except for an operation for spinal and hand trauma years before. This patient had concomitant ischaemic coronary artery disease and aortic stenosis. The identification of T whippelii infection in such a patient is in agreement with reports of cases of T whippelii infective endocarditis that lack overt gastrointestinal disease.20 As the patient had had no extracardiac symptoms, a diagnostic work up for Whipple’s disease was not undertaken.
Patient 7 had a non-responsive infective endocarditis episode complicated by heart failure. Positive specific antibodies against Borrelia species led us to perform a specific assay for B burgdorferi which was positive from a valve tissue sample. The assay identified the variant sensu lato, the causal agent of Lyme disease in some areas of Europe and the USA. Although it has been reported that non-borrelial subacute infective endocarditis may sometimes cause cross reactivity and lead to seropositivity to anti-borrelia antigens,31 PCR confirmed the presence of B burgdorferi sensu lato in valve tissue in this particular patient. There are, to our knowledge, no reports of Borrelia species causing infective endocarditis, so the pathogenic role of B burgdorferi in this case needs to be further investigated. Techniques such as in situ PCR and immunohistochemistry will help clarify the significance of the finding.
The only case of fungal native infective endocarditis corresponded to a kidney transplant patient (case 3) with concurrent disseminated aspergillosis. The diagnosis was confirmed by histopathology of the resected valve.
The microbiological spectrum of cases of prosthetic infective endocarditis in this study is similar to that shown in several other studies,1 with exception of one patient who developed an early PVE episode (less than two months after the operation) and in whom Streptococcus species was identified by PCR. As the prevalence of early streptococcal PVE seems to be very low (about 2%), the above finding is unusual although possible.29 Fungal infective endocarditis episodes caused by Candida species and Aspergillus species (patients 1 and 11, respectively), are often identified in patients with prosthetic valves.11
Overall, UNB and UNF systems detected and identified the aetiology of infective endocarditis in 11 of 15 patients (73.3%). In five cases, identification reached the species level and in the remaining six the genus level. These figures should be acceptable as a starting point for optimisation of the method. There are no published data to compare with our results, except for a few cases in the study by Goldenberger and colleagues,8 in which four culture negative cases of infective endocarditis were correctly detected and identified by PCR and further sequencing. We have not used sequencing as a standard procedure but we agree that 16S rRNA AFLP-RFLP analysis alone might not be sufficient to allow species level discrimination in a proportion of bacterial infective endocarditis episodes. In cases of fungal infection, several studies have shown that species level identification can be achieved by AFLP-RFLP in most cases, although sequencing may be necessary in a few.23,24
Although all possible measures were used to avoid carryover and cross contamination (dUTP, uracil-N-glycosylase, 8-methoxy-psoralen, and ultraviolet irradiation27,28,32–36), in three patients (20%) it was not possible to identify the pathogen because of a strongly contaminating background which did not allow further sequencing. Contamination is therefore one of the most important problems when dealing with molecular detection systems. For this reason, the need for aseptic conditions during sample collection and storage, as well as in the molecular biology laboratory, cannot be overemphasised.37,38
Aside from the problems inherent in this technology, our study shows that molecular detection of pathogens is a promising tool for clinical use. The fact that quantitative or semiquantitative results can be delivered within a few hours after collecting the samples is, on its own, a good reason to support the use of this technology in selected diseases and groups of patients.4,39 Moreover, the possibility of quantitation by real time PCR makes the technology even more attractive. This advance would eliminate the need for gel electrophoresis for amplicon detection and would render PCR faster and more accurate. Positive samples after real time PCR can be rapidly sequenced and a report issued within a few hours. In addition, recent studies showing the possibility of characterising bacterial resistance by molecular methods in cases of infective endocarditis40 would, once validated, add a very useful clinical dimension to the currently available spectrum of molecular techniques in infectious diseases.
Conclusions
PCR based methods are highly sensitive, reliable, and rapid tools for the microbiological diagnosis of culture negative infective endocarditis. The clinical applicability of universal bacterial and fungal molecular detection systems is, in our opinion, feasible as long as adequate validation and controls are used in routine analyses. Constant surveillance for detecting and eliminating carryover and cross contamination, along with adequate clinical selection of cases, would be the best approach to take advantage of PCR based detection of pathogens in culture negative infective endocarditis.
Acknowledgments
This work was supported in part by grant NM6780-3/2001 from the Internal Czech Grants Agency of the Ministry of Health of the Czech Republic. Many thanks to Dr David P Nelson for comments and review of English usage.