Article Text

Abstract
Objective: The prevalence and types of various cardiovascular diseases in different age groups as well as the outcomes of cardiac surgery and other interventions were assessed in a population of 75 Williams syndrome (WS) patients aged 4 months to 76 years (median 22.7 years).
Study design: The diagnosis of WS was in each case confirmed by the clinical phenotype and by a FISH test showing elastin hemizygosity. Clinical and operative data were collected from all hospitals where the patients had been treated.
Results: Cardiovascular symptoms were evident in 35 of 75 (47%) WS children at birth. During follow up, 44 of 75 (53%) WS patients were found to have cardiovascular defects. Among them, the definitive diagnosis was made before 1 year of age in 23 (52%) infants, between 1 year and 15 years of age in 14 (32%) children, and older than 15 years of age in 7 (16%) adults. Multiple obstructive cardiovascular diseases were found in six infants. Supravalvular aortic stenosis (SVAS) was diagnosed in 32/44 (73%), pulmonary arterial stenosis (PAS) in 18/44 (41%), aortic or mitral valve defect in 5/44 (11 %) of cases, and tetralogy of Fallot in one (2%) case. Altogether, 17/44 (39 %) underwent surgery or intervention. Surgery was most frequently performed in the infant group (6% v 21% v 0%, p=0.004). After 1 year of age, seven patients underwent SVAS relief and two cases PAS relief. Postoperatively there was no mortality (median follow up time 6.9 years). Arterial hypertension was found in 55% of adults. In three adults, arterial vasculopathy was not diagnosed until necropsy.
Conclusions: Our data indicate the following in WS. Cardiac symptoms are common in neonates. Heart disease diagnosed in infancy frequently requires operation. After 1 year of age, PAS tends to improve and SVAS to progress. Life long cardiac follow up is necessary because of the risks of developing vasculopathy or arterial hypertension.
- Williams syndrome
- elastin vasculopathy
- cardiovascular manifestations
- supravalvular aortic stenosis
- ASD, atrial septal defect
- CMP, cardiomyopathy
- CoA, coarctation of the aorta
- LPA, left branch of pulmonary artery
- MPA, main pulmonary artery
- PAS, pulmonary arterial stenosis
- RPA, right branch of pulmonary artery
- SVAS, supravalvular aortic stenosis
- VSD, ventricular septal defect
- TOF, tetralogy of Fallot
- AS, aortic stenosis
- AI, aortic insufficiency
- MS, mitral stenosis
- MI, mitral insufficiency
- WS, Williams syndrome
Statistics from Altmetric.com
- ASD, atrial septal defect
- CMP, cardiomyopathy
- CoA, coarctation of the aorta
- LPA, left branch of pulmonary artery
- MPA, main pulmonary artery
- PAS, pulmonary arterial stenosis
- RPA, right branch of pulmonary artery
- SVAS, supravalvular aortic stenosis
- VSD, ventricular septal defect
- TOF, tetralogy of Fallot
- AS, aortic stenosis
- AI, aortic insufficiency
- MS, mitral stenosis
- MI, mitral insufficiency
- WS, Williams syndrome
Williams syndrome (WS) is a complex developmental disorder comprising cardiovascular disorders, mental retardation with a peculiar cognitive profile, dysmorphic facies and body features, and occasional hypercalcaemia.1 It is associated with a microdeletion in the chromosomal region 7q11.23 encompassing, among others, the elastin gene. The syndrome is routinely confirmed by detecting elastin hemizygosity by fluorescence in situ hybridisation (FISH).2–4 Previous reports suggest that hemizygosity of the elastin gene is responsible for the typical vasculopathy of WS, namely supravalvular aortic stenosis (SVAS) and pulmonary arterial stenosis (PAS).4,5 Deficient elastin may also account for other frequent connective tissue abnormalities in WS, such as hernias, lax skin, the impression of premature aging, stiffness of joints, and may even be the origin of vascular hypertension.
The pathogenic mechanism underlying the WS vasculopathy is not yet understood. Reduced and abnormal elastin content in the media of developing vessels may lead to recurrent injury and fibrosis. Vascular inelasticity may increase haemodynamic stress to the endothelium leading to intimal proliferation of smooth muscle and fibroblasts, fibrosis, and luminal narrowing of the vessels.4 In clinical studies, it has been shown that with time PAS tends to improve and SVAS to progress.6 Failure of growth at the sinotubular junction might be responsible for the progression of the aortic lesions.6,7 Obstructions may also occur in the aortic arch, the innominate and carotid arteries, and other major aortic side branches.8,9 In addition, a localised coarctation is encountered in patients with SVAS.10 Apart from obstructive lesions caused by arterial wall thickening, WS patients also have other left sided anomalies, namely mitral and aortic valve anomalies.11
The aims of the present study were to assess the prevalence and the types of cardiovascular conditions as well as the ages of manifestation of symptoms in a population of 75 well documented WS patients. In addition, the overall outcome of the different cardiovascular diseases was evaluated.
PATIENTS AND METHODS
This retrospective follow up study consists of 75 patients with WS with a median age of 22.7 years (range 0.3-76 years) (table 1). The diagnoses of WS were made in the Hospital for Children and Adolescents in Helsinki and in the Department of Medical Genetics, the Family Federation of Finland between the years 1974 and 2000. In each case, the diagnosis was confirmed by the clinical phenotype assessed by an experienced medical geneticist and the typical elastin gene hemizygosity shown by FISH. The study was approved by an institutional review committee.
Clinical characteristics of the study population (n=75)
The patients’ neonatal well being was assessed by reviewing their hospital records for gestational age, birth weight, birth length, Apgar scores at 1 and 5 minutes as well as hyperbilirubinaemia, hypoglycaemia, respiratory problems, septic infections, thrombocytopenia and hypercalcaemia, neurological hyperexcitability, and failure to thrive. Cardiac symptoms were defined as murmur, heart failure, absent femoral pulses, or cyanosis.
Clinical cardiovascular data were collected from all the hospitals where the patients had been treated. Included are the data on the age and type of the cardiac symptoms, ECG, chest x ray, echocardiography, and heart catheterisation findings. Cardiac ultrasound by a paediatric or adult cardiologist was performed in 65/75 patients (87%). Heart catheterisation was performed in 19/75 patients (25%). The types of cardiovascular defects found were supravalvular aortic stenosis (SVAS), pulmonary arterial stenosis (PAS), aortic coarctation (CoA), cardiomyopathy (CMP), tetralogy of Fallot (TOF), aortic valve defect (aortic stenosis (AS) or aortic insufficiency (AI)), and mitral valve defect (mitral stenosis (MS) or mitral insufficiency (MI)). SVAS was diagnosed by echocardiography or catheterisation as a localised or diffuse narrowing of the ascending aorta associated with a pressure gradient of more than 10 mm Hg. PAS was diagnosed as a local or diffuse narrowing of the main pulmonary artery or main branches with a pressure gradient of more than 10 mm Hg. All pressure gradients in vascular stenosis before and after balloon dilatation or surgery were registered. Cardiac procedures were defined as heart surgery or cardiac intervention (angioplasty by balloon dilatation). Surgical technique and the postoperative outcome were reviewed.
The diagnosis of arterial hypertension was made using age and gender specific normal data. In repeated measurements, mean daytime ambulatory BP of more than or equal to 135/85 mm Hg was classified as hypertensive. Four limb measurements were used in 10 adult patients (>15 years) and diurnal blood pressure pattern in five patients. All the medication the patients had used was recorded. In the follow up, patient records were reviewed to assess associated cardiovascular diseases like ischaemic heart disease. Updated clinical information was collected from the hospitals and the patients, their family members, or other caretakers. The most recent blood pressure, echocardiography, and x ray data were reviewed. Information on the macroscopic and histological postmortem examinations was gathered.
The data are presented as means (SD) or as median and range. Dichotomous variables were contrasted by using chi-squared analysis (one tailed) or by t test (two tailed). Continuous variables were analysed by t test; p<0.05 was considered significant.
RESULTS
In the newborn period, cardiovascular symptoms were evident in 35 of 75 (47%) WS children, of whom 28 had cardiac murmur, four had heart failure, two had cyanosis, and one had absent femoral pulses (table 1). Among 35 symptomatic newborns, 27 (77%) were found to have a structural heart defect. During the follow up, overall 44 (59%) of the 75 WS patients were diagnosed as having a structural heart defect or vascular disease. Based on the age at the definitive diagnosis of cardiovascular defect, the study population was classified into three groups: (1) infants (n=23), that is, diagnosis made before 1 year of age, (2) children (n=14), that is, diagnosis made between 1 and 15 years of age, and (3) adults (n=7), that is, diagnosis made older than 15 years of age (table 2).
The age at diagnosis and need for cardiac intervention in different heart defects (n=44)
A total of 14 of 23 (61%) patients in the infant group needed surgery or intervention. Five infants were shown to have the most complicated elastin vasculopathy: SVAS accompanied by PAS and hypoplastic aortic arch with localised CoA and hypertrophic CMP. Four of them manifested heart failure and were operated on before 6 months of age (table 3). In addition, four patients were diagnosed as having SVAS and were later operated on at the age of 3, 9, 10, and 17 years of age, respectively. Two other patients with SVAS and PAS had cardiac procedures at the ages of 0.5 and 6 years, respectively. In addition, three patients with PAS had cardiac procedures at the ages of 0.4, 0.6, and 2.5 years. One child with tetralogy of Fallot had total corrective heart surgery at the age of 1.3 years (table 3).
The outcome of cardiac intervention in patients with Williams syndrome (n=17)
In the infant group, there were two deaths (9%). One girl with moderate SVAS, PAS, moderate CoA, and hypertrophic CMP was followed without operation and died at the age of 4 years of pneumonia. In addition, one baby boy died suddenly of heart failure at the age of 4 months without diagnosis. Postmortem study showed a severe hypertrophic CMP and an abnormally thick aortic wall but no visible obstructions in the main arteries. These postmortem findings raised the suspicion of WS which was then confirmed by FISH.
In the children’s group, SVAS was the most frequent diagnosis (79%). Three of 11 (27%) patients had cardiac operations at the ages of 8, 11, and 45 years, respectively. In patients with PAS (n=2) or mitral valve insufficiency (n=1) no cardiac procedures had been performed (table 2).
As many as seven (16%) WS patients were diagnosed with a cardiovascular defect after 15 years of age. No cardiac procedures had been necessary in this adult group. Two patients with aortic stenosis and one male patient with mitral valve defect remain in follow up. Of the three patients with SVAS, two were not diagnosed until necropsy. In addition, one man with arterial hypertension died at the age of 76 years, the cause of his rapidly declining condition remaining obscure. Necropsy showed SVAS with hypoplastic aortic arch and severe mitral valve defect with hypoplastic papillary muscles.
The diagnosis of SVAS with or without associated defect was made in 32 of 44 patients (73%). Cardiac interventions were performed in 13 patients (41%) (table 3). All of them had preoperative heart catheterisation and the mean preoperative pressure gradient ranged from 30 to 150 mm Hg (77 mm Hg). In patients without intervention (n=18), mean pressure gradient measured by cardiac ultrasound was 17 mm Hg (12-26 mm Hg) with a median follow up age of 9.6 years (range 2-41 years).
PAS was diagnosed in 18 of 44 (41%) patients with cardiac defect; all cases were diagnosed before 15 years of age. Altogether, 10 of the 18 (56%) patients with PAS needed cardiac intervention (table 3). Before intervention, mean pressure gradient was 45 mm Hg (range 20-90 mm Hg). In patients without intervention (n=8), mean pressure gradient measured by ultrasound was 24 mm Hg (11-50 mm Hg) with a median follow up age of 7 years (range 2-20 years).
The type of cardiac procedures and their outcomes are presented in table 3. The median follow up period after surgery or intervention was 6.9 years (range 1-15 years). In the infant group, cardiac surgery or intervention were more frequently needed than in the children’s or adult groups (61%, 21%, and 0%, p=0.004). There was no mortality. After aortoplasty or aortic angioplasty, mild to moderate aortic valve insufficiency was found in four cases. In addition, mild restenosis was found in three cases. In children with CoA, mild to moderate recoarctation was found in all three operated cases. After pulmonary artery reconstruction, two children had restenosis.
The outcome of the whole study population is presented in fig 1. Arterial hypertension was found in 23 of 42 patients (55%) older than 15 years. During the monitoring period, antihypertensive medications were used by 14 of 23 (61%) patients with hypertension. These included: beta blocker only (n=6), beta blocker and diuretics (n=2), calcium blocker (n=2), ACE blocker (n=2), and diuretics only (n=2).
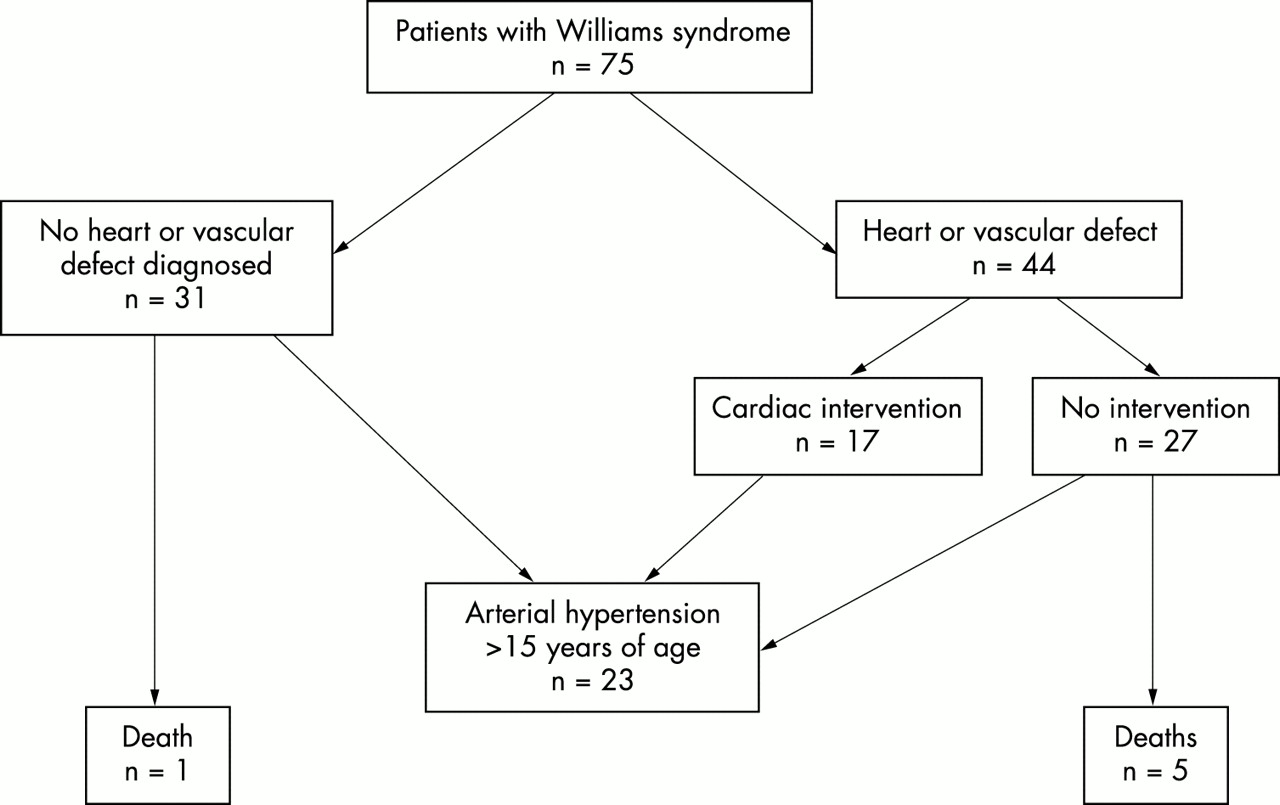
{kind=link}
The clinical outcome of 75 patients with Williams syndrome.
In the whole study population, there were six deaths (8%), of whom five patients had arterial vasculopathy diagnosed either by ultrasound or necropsy (table 2). In addition, one female patient with arterial hypertension associated with coronary artery disease and diabetes died at the age of 66 years. No necropsy was performed.
DISCUSSION
Our study shows that 77% of those with cardiac symptoms as a newborn were found to have arterial vasculopathy or intracardiac defect. Therefore, cardiac symptoms during the neonatal period are typical of children with WS and might aid in making the diagnosis of WS when other characteristic features can remain unrecognised.
We have shown that heart or vascular disease occurred in 53% of patients with WS aged from newborn to 76 years and in 52% of cases the diagnosis was completed before 12 months of age. Six infants aged 0 to 4 months had the most widespread cardiovascular involvement, including the ascending and descending aorta, pulmonary arteries, or myocardium. Among these youngest children, the prognosis was poor; two died suddenly and four needed cardiac intervention. In a recent case report, a 30 week old fetus with hydrops and a chromosomal translocation breakpoint at 7q11.23 was shown to have narrow aorta and pulmonary arteries.12 The baby died shortly after delivery and necropsy showed diffuse tubular thickening with luminal narrowing of the aorta, aortic branches, and the pulmonary arteries.12 Bird et al10 described 10 WS children of 1 month to 6 years of age who died suddenly. In seven cases, the necropsy findings showed coronary artery stenosis and severe biventricular outflow tract obstruction. The possible mechanisms implied that sudden death was the result of myocardial ischaemia, decreased cardiac output, or arrhythmia.10 Our two cases of sudden death had severe CMP; one died at the age of 4 months and the other at 4 years. Myocardial ischaemia owing to coronary artery stenosis was not found. However, a drop in cardiac output can result in coronary insufficiency. A ventricular arrhythmia generated by an ischaemic myocardium is a possible mechanism to explain sudden death in these cases.10,13–15
Cardiac interventions were most frequently required in the infant group. Dilatation of the pulmonary arteries was most often, in five cases, necessary. Aortic coarctation relief was performed in three cases and SVAS repair in three cases. In children over 1 year of age, dilatation of PAS was required in only two cases and two patients with PAS showed spontaneous recovery. However, SVAS relief in this group was more frequently necessary (seven cases). In addition, 19 patients with SVAS aged from 2 to 25 years are under strict follow up. These findings support the results of a previous study of Kim et al,6 which showed that, with time, PAS tends to improve spontaneously and SVAS to progress. Whether PAS diagnosed in infancy might be followed without intervention remains to be resolved in future studies.
In our study population, there were four cases with an unusually severe WS phenotype and a large deletion in 7q11.23 visible on a prometaphase chromosome study. In the case report of Wu et al,16 a female patient was described with a similar large deletion and a moderately severe SVAS. Common to our patients were severe developmental retardation, epilepsy, joint stiffness and contractures, scoliosis, small chin, prominent metopic suture, small deciduous teeth, and frequent chest and ear infections. The cardiovascular manifestations varied. A pair of male twins was operated on; one twin had reconstruction of pulmonary arteries and closure of an ASD and the other had a total correction of tetralogy of Fallot. A third patient, a female, died at the age of 4 years because of bilateral outflow tract obstruction and hypertrophic CMP. The fourth patient is an adult female with no findings of a heart defect so far.
In general, as shown by many surgical follow up studies, aortic arch surgery can be performed safely and with excellent early and late results in children with SVAS.9,17–19 Stamm et al18 showed that in the most severe form of elastin arteriopathy surgical treatment was palliative, but in conjunction with balloon dilatation it offers good long term survival to patients with WS. In their series of 33 WS patients, there were six early deaths and one late death. In our series of 17 patients, there was no mortality. In addition, during the follow up, there have been no late deaths. Mild restenosis shown by ultrasound was found in eight patients after surgical relief of SVAS, PAS, or CoA. One of them had a reintervention, balloon dilatation of recoarctation. Mild to moderate aortic regurgitation was found in four patients after SVAS relief. At follow up, altogether five operated patients are taking cardiovascular medication, digitalis in three and a beta blocker in two patients. Therefore, compared with earlier studies,9,18,19 the total outcome of the operated WS patients with the median follow up time of 6.9 years was relatively good.
Severe mitral regurgitation in WS patients requiring surgical treatment at the ages of 8 and 11 years was described by Maisuls et al.20 In our patients, no mitral valvulotomy or prosthesis has been performed. One child had mitral valve prolapse diagnosed at the age of 6 years. So far, no medication is being administered. Furthermore, one adult female with mitral regurgitation has been followed up since 34 years of age. She has medication for arterial hypertension. In addition, in one adult male patient, the diagnosis of severe mitral valve defect was made at necropsy.
In our study, we had three adult patients aged 26, 51, and 76 years in whom the diagnosis of elastin arteriopathy was made at necropsy. All these adults had cardiac symptoms before death. However, arteriopathy was not diagnosed. This finding is similar to earlier case reports and shows that SVAS can be difficult to diagnose without cardiac catheterisation in adults.14,21,22 Two of these adults died of severe systemic infection. It is obvious that the hypertrophic or dilated myocardium as a result of vasculopathy was not capable of maintaining systemic cardiac output under severe haemodynamic strain.14,22
Broder et al23 studied blood pressure in 20 WS patients using 24 hour ambulatory monitoring. They found that hypertension defined by raised mean daytime blood pressure was present in 40% of WS patients versus 14% of controls. In our study, arterial hypertension was found in 55% of adult patients and among them antihypertensive medication was used in 61% of patients. Therefore, all subjects with WS require careful blood pressure monitoring. Recordings should be followed with regular measurements and also with 24 hour ambulatory monitoring when necessary. Because of the potential possibility of distal vascular stenosis, four limb measurements are preferred at follow up.
Limitations of the study
Any retrospective study has inherent limitations. The study took place over multiple decades and the diagnostic and surgical technology, in addition to the medication, has changed substantially over the study period. Cardiac ultrasound was available from 1980 and before that cardiac catheterisation was the only trustworthy method of diagnosing cardiovascular lesions. However, all our WS patients were diagnosed and followed up by a paediatric or adult cardiologist. Any patient with insufficient records was excluded.
CONCLUSION
In conclusion, our data show that in patients with WS cardiac symptoms in the newborn period are common. Since WS easily remains unrecognised in newborns, we recommend that all dysmorphic babies be examined by cardiac ultrasound. When elastin arteriopathy is diagnosed as an infant, the prognosis is poorest. There is a high risk of sudden death. In addition, cardiac interventions, including dilatations of both great arteries, are frequently required. After 1 year of age, PAS tends to improve spontaneously and SVAS to become worse. When cardiac interventions were used, the prognosis was relatively good and no operative mortality was found. In some adult patients with WS, cardiac catheterisation may be necessary to diagnose elastin arteriopathy. It is noteworthy that a considerable number of the WS associated cardiovascular problems including SVAS may not manifest until adult age and the symptoms might be missing or non-specific, thus hampering diagnostic procedures and proper treatment. Therefore, all WS patients with or without symptoms, operated on or not, should be followed throughout life by a cardiologist.
Acknowledgments
Financial support for this study was received from the Rinnekoti Foundation.