Article Text

Statistics from Altmetric.com
Following coronary stent placement, platelet activation is a major determinant of the risk of subacute stent thrombosis.1 Combined antiplatelet treatment with ticlopidine and aspirin reduced platelet activation after coronary stenting1. Although combined antiplatelet treatment consisting of aspirin and ticlopidine has significantly reduced early ischaemic events following coronary stenting, stent thrombosis still occurs in up to 1% of treated patients, especially in the early days after the intervention, probably because of delayed onset of action of ticlopidine. Clopidogrel is a ticlopidine-like novel thienopyridine inhibitor of ADP induced platelet activation.2 Clopidogrel differs from ticlopidine in that it has a favourable safety profile compared to ticlopidine and reveals an accelerated antiplatelet activity after first administration. The present study sought to investigate the antiplatelet effect of various doses of clopidogrel in patients undergoing coronary stent placement; comparison was made with standard ticlopidine treatment.
Thirty patients were randomised into three treatment arms: group I (n = 10), ticlopidine 2 × 500 g as loading dose and 2 × 250 mg daily thereafter; group II (n = 10), clopidogrel 1 × 300 mg loading dose and 1 × 75 mg per day; or group III (n = 10), clopidogrel 1 × 600 mg plus 2 × 75 mg daily thereafter. All patients received aspirin 2 × 100 mg per day concomitantly. Peripheral venous blood samples were taken with a loose tourniquet through a short venous catheter inserted into a forearm vein before and then 2, 4, 24, and 48 hours after first administration of antiplatelet medication. Platelet aggregation in response to ADP (5 or 20 μmol/l) and to TRAP (25 μmol/l) was evaluated by optical aggregometry in citrated blood samples,3 and ATP release as marker for dense granule secretion was determined by luminometry.3 Surface expression of P-selectin (CD 62P binding) was analysed by flow cytometry according to published methods and served as index of degranulation of α-granules.3
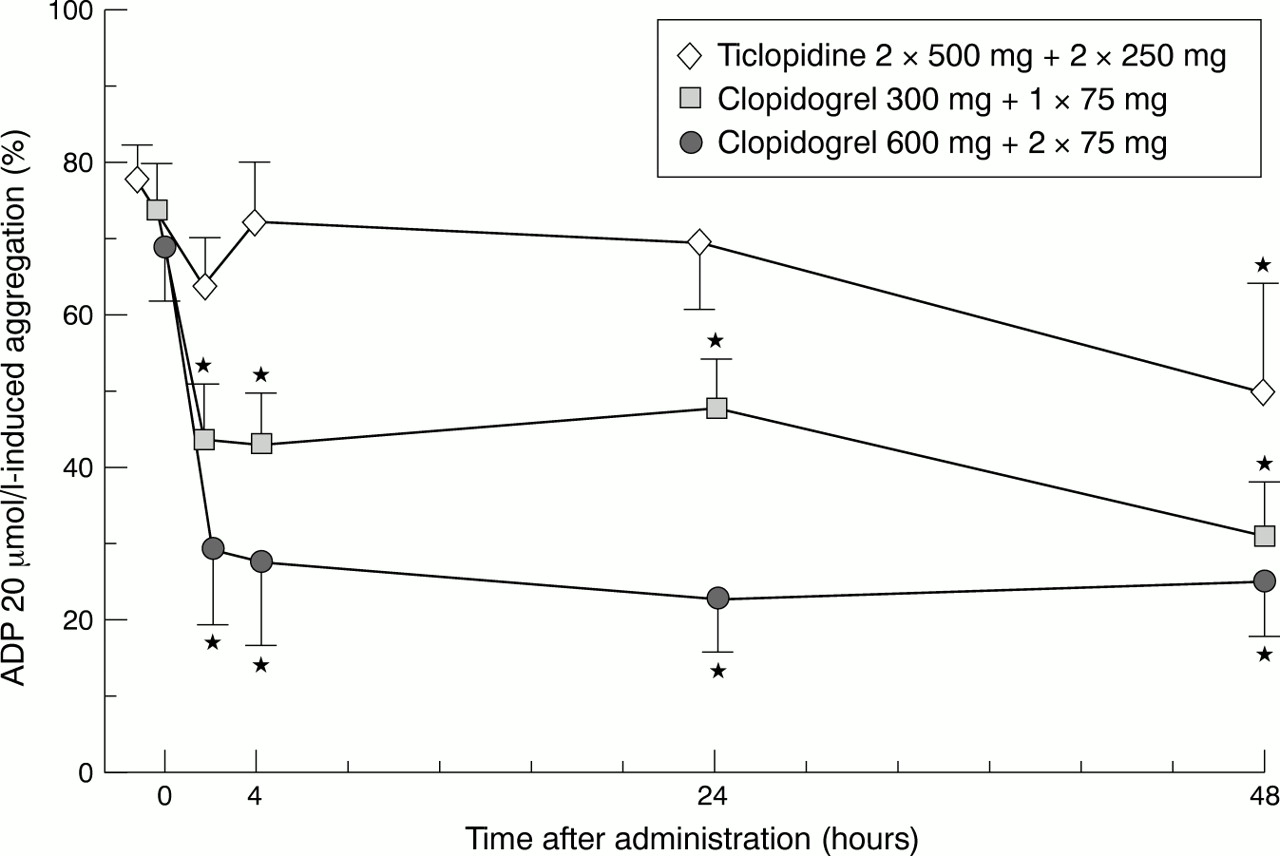
Compared to baseline values ADP (20 μmol/l) induced platelet aggregation was inhibited significantly by approximately 55–59% (p < 0.01) within the first 4 hours after drug administration in patients receiving the 600 mg loading dose (fig 1). Substantially less inhibition of ADP (20 μmol/l) induced aggregation (38–40%) was found in patients receiving an initial loading dose of clopidogrel of 300 mg compared to the patient group treated initially with 600 mg (p < 0.01) (fig 1). No substantial platelet inhibition within the early hours after drug administration was observed in patients treated with ticlopidine (fig 1). Inhibition of ADP (20 μmol/l) induced platelet aggregation further decreased after 48 hours in patients receiving 300 mg clopidogrel (up to 52%), whereas in patients treated with 600 mg clopidogrel platelet aggregation remained on the level of inhibition found during the first few hours after drug administration (fig 1). In patients treated with ticlopidine, ADP (20 μmol/l) induced platelet aggregation started to be substantially inhibited by approximately 38% at 48 hours (p < 0.01) (fig 1). Similar results were obtained for all three groups of patients when a weaker stimulus of platelet aggregation was applied (ADP 5 μM) (data not shown).

{kind=link}
Effect of high loading dose of clopidogrel on ADP induced platelet aggregation. Patients were randomised into three treatment arms: ticlopidine 2 × 500 mg plus 2 × 250 mg daily thereafter (n = 10); clopidogrel 300 mg loading dose plus 1 × 75 mg daily (n = 10); and clopidogrel 600 mg loading dose plus 2 × 75 mg daily thereafter (n = 10). All patients received aspirin 2 × 100 mg/day concomitantly. Platelet aggregation was studied after stimulation with ADP (20 μmol/l) by light transmittance aggregometry in citrated platelet rich plasma. *Significant difference (p < 0.05) compared to starting concentrations.
When the thrombin related activating peptide (TRAP), a strong agonist of platelet aggregation, was used to stimulate platelets, only weak inhibition of platelet aggregation was observed in all three patient groups throughout the observation time. Inhibition of TRAP (25 μM) induced platelet aggregation at 2 hours and 48 hours was 11% and 8%, respectively, (p < 0.05) in the group treated with 600 mg clopidogrel, and 5% and 9% (p < 0.05), respectively, in the group treated with 300 mg clopidogrel. In patients receiving ticlopidine, TRAP induced aggregation tended to be slightly decreased by 5% after 2 hours or 48 hours; however, this did not reach a significant level (data not shown).
A substantial decrease of ADP (20 μM) induced α-degranulation, as evaluated by the surface expression of P-selectin (CD62P), was exclusively found in patients treated with 600 mg clopidogrel loading dose. The median (25% and 75% quartile) of the mean intensity of immunofluorescence (MIF) of CD62P was 244 (231, 261) before and 207 (183, 223) 48 hours after administration (p < 0.05) (data not shown). In the group treated with 300 mg clopidogrel or with ticlopidine no substantially inhibitory effect on α-degranulation was seen throughout the observation time (data not shown). Furthermore, in none of the treatment groups was TRAP (25 μM) induced α-degranulation substantially inhibited (not shown). In none of the groups of patients was substantial inhibition of ADP (5 and 20 μM) or TRAP (25μM) induced ATP release found (data not shown).
The present study shows that a combined antiplatelet treatment with aspirin and clopidogrel in a high loading dose of 600 mg and a continuous dose of 150 mg per day is superior to the combination of aspirin and standard clopidogrel (300 mg loading dose plus 75 mg per day)4 or ticlopidine (2 × 500 mg loading dose plus 2 × 250 mg per day) treatment1 in suppressing platelet aggregation after coronary stenting. Administration of 600 mg clopidogrel accelerates inhibition of ADP induced platelet aggregation during the first few hours to a level that cannot be accomplished by the conventional dosing regimen of 300 mg clopidogrel4 ,5before 48 hours after first drug administration. The accelerated effect of 600 mg clopidogrel narrows the therapeutic gap of ticlopidine and might be beneficial, especially for high risk patients treated with coronary stenting. The study shows furthermore that clopidogrel in a high loading dose of 600 mg is able to reduce the degranulation of α-granules. Since platelet release products derived from α-granules are mitogenic, it is possible that the combination of aspirin and clopidogrel in a high dosage might modulate platelet dependent restenotic processes following coronary angioplasty. On the other hand, the lack of inhibition of aggregation and degranulation of TRAP stimulated platelets in all three treatment groups indicates that even combined antiplatelet treatment with aspirin and clopidogrel cannot substantially reduce platelet activation in an environment of high thrombin activity. This might be of importance in patients with acute coronary syndromes or who are being treated by fibrinolysis for acute myocardial infarction. It is tempting to speculate that in these patients the addition of further antithrombotic compounds including glycoprotein IIb/IIIa blockers and antithrombins to aspirin and clopidogrel might favour a better clinical outcome.