Article Text

Statistics from Altmetric.com
- AKT: cellular homologue of transforming oncogene of AKT8 retrovirus
- ERK: extracellular signal related kinase
- FLIP: Fas-like inhibitory protein
- IAP: inhibitor of apoptosis protein
- SAPK: stress activated protein kinase
- TNF: tumour necrosis factor
- TUNEL: terminal UTP nick end labelling
- VSMC: vascular smooth muscle cell
Apoptosis, or programmed cell death, is a process through which multicellular organisms dispose of cells efficiently. Much has been discovered about the molecular control of apoptosis since its initial description as a series of morphological events.1 Apoptosis defines a type of cell death distinct from the more conventional necrotic death, seen classically in myocardial infarction, on the basis of characteristic morphological features (table 1, fig 1). Although these descriptions and distinctions are useful, there is a great deal of overlap between apoptosis and necrosis in morphological features and biochemical events. Indeed, apoptosis is frequently followed by secondary necrosis of cells, especially if there is failure of clearance or ingestion of apoptotic bodies.
Characteristic features of apoptosis versus necrosis

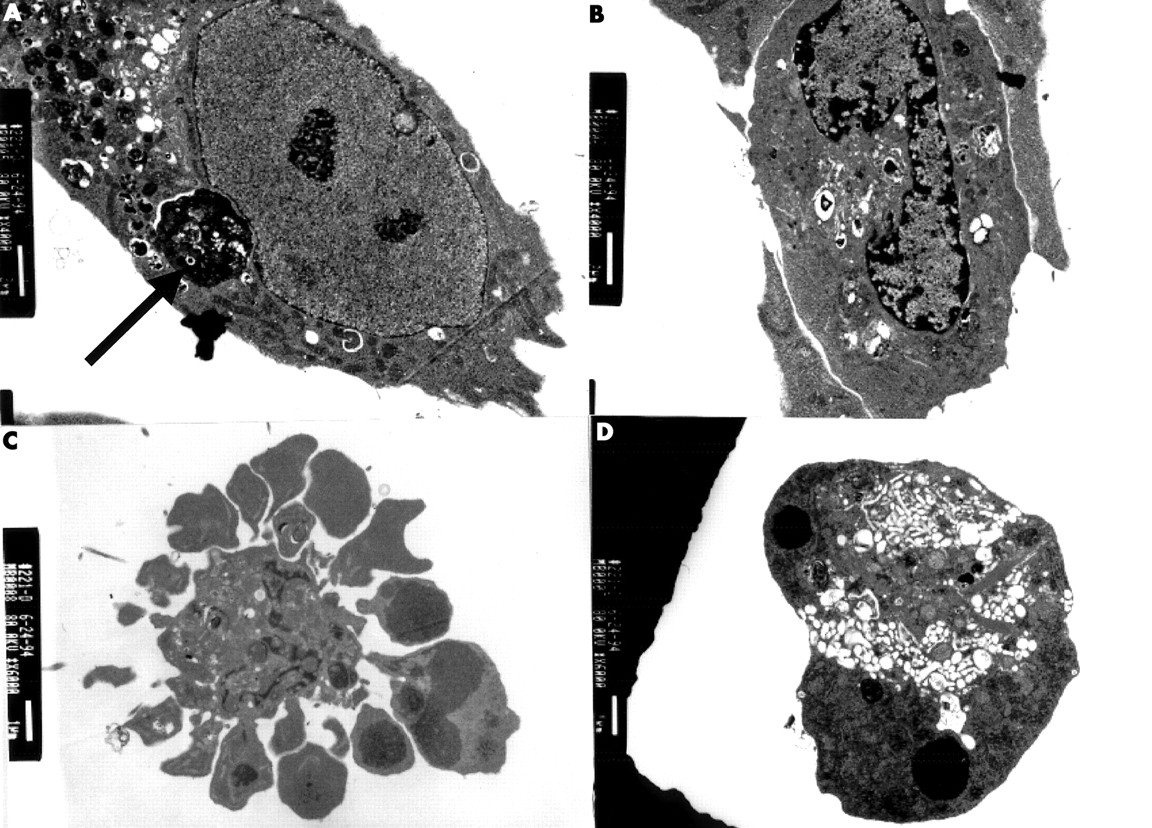
Electron microscopic appearances of a human vascular smooth muscle cell (VSMC) undergoing apoptosis in culture. (A) Normal appearance of a human VSMC. VSMC also contains an apoptotic body (arrow). (B) Peripheral condensation of nuclear chromatin. (C) Intense membrane blebbing and vesicle formation in apoptosis, with condensation of the nuclear chromatin into clumps. (D) An apoptotic body, the end product of apoptosis.
DETECTION OF APOPTOSIS
Apoptotic cells undergo a characteristic cascade of biochemical events (see Regulation of apoptosis), many of which are useful in detecting apoptotic cells. In particular, apoptotic cells expose specific membrane phospholipids that can be detected with labelled marker proteins (for example, phosphatidylserine detected with fluorescently labelled annexin V) and cleave their DNA into specific fragments that are the basis for the enzyme linked assays to detect fragmented DNA (for example, terminal UTP nick end labelling, or TUNEL). Biochemical signalling during apoptosis, such as activation or cleavage of specific caspase enzymes (see below) can also be used on both cells and tissue samples. While helpful, the gold standard for detecting apoptosis is still based on morphology at both the light and particularly the electron microscopic level, where the features outlined in table 1 are easily distinguished.
APOPTOSIS IN THE HEART
The adult cardiomyocyte has limited (if any) ability to proliferate. Correspondingly, apoptosis is observed infrequently in adult hearts. In contrast, cardiomyocyte apoptosis plays a critical role in heart formation, such as formation of septa between cardiac chambers and valves. This evidence suggests that defects in apoptosis can result in congenital heart disease. Major foci of apoptosis include zones of fusion of the atrioventricular or bulbar cushions, and both aortic and pulmonary valves in non-myocytes. Myocyte apoptosis also occurs in the interventricular septum and right ventricular wall after birth, during the transition from fetal to adult circulations. The conducting tissue also undergoes apoptosis, and aberrant apoptosis is implicated in congenital heart block and long QT syndrome or the persistence of accessory pathways.
Apoptosis in ischaemia/infarction
Ischaemia is a potent inducer of both myocyte necrosis and apoptosis in vitro. Thus, deprivation of oxygen alone, or in addition to serum withdrawal and deprivation of glucose, induces neonatal myocyte apoptosis. Ischaemia alone can induce apoptosis in the ischaemic territory in vivo, and this may be reduced by reperfusion. However, although reperfusion may limit ischaemia induced apoptosis, reperfusion itself may accelerate the appearance of apoptosis in the reperfused regions.2
Myocardial infarction has been considered to be a prime example of necrotic cell death, because of the breakdown of cellular energy metabolism. However, apoptosis of cardiomyocytes also occurs in a temporally and spatially specific manner. Thus, acute myocardial infarction manifests both forms of cell death,3 with apoptosis particularly occurring at the hypoperfused “border” zones, between a central area of necrosis and viable myocardium. The central, unperfused region4 also manifests apoptosis, particularly within the first six hours, although between 6–24 hours necrosis is more common. Apoptosis in the remote non-infarcted myocardium may be partly responsible for myocardial remodelling and dilatation after myocardial infarction, and may be amenable to treatment.
Apoptosis in heart failure
The finding that cardiomyocyte apoptosis occurs in the end stage human heart indicates that apoptosis may contribute to heart failure in a variety of situations.5, 16 Aging is associated with myocardial cell loss, and cardiomyocyte apoptosis may be the mechanism of the gradual deterioration in cardiac function. In humans undergoing transplantation, apoptosis can be observed,6 with some studies suggesting higher levels in ischaemic versus idiopathic dilated cardiomyopathy.5 The transition from compensated to decompensated hypertrophy is also associated with myocyte apoptosis in animals,7 and high levels of apoptosis are seen in arrhythmogenic right ventricular dysplasia, a condition characterised by myocardial replacement with fibrofatty material. Finally, there is increasing evidence that toxic cardiomyopathies, such as that induced by doxorubicin (Adriamycin), are associated with cardiomyocyte apoptosis.
Although the evidence that apoptosis promotes heart failure is persuasive, the present problem is defining by what extent. Vastly different rates of apoptosis have been reported in both human and animal heart failure, with rates of up to 35.5%.5 While these death rates may be seen only in very localised areas, given that apoptosis takes less than 24 hours to complete, such rates would result in rapid involution of the heart. More recently, rates of < 0.5% have been consistently reported in end stage heart failure, which make far more physiological sense. In addition, in end stage heart failure necrosis is still (up to seven times) more frequent than apoptosis.
APOPTOSIS IN THE VESSEL WALL
Vascular smooth muscle cells (VSMCs) within the vessel wall can both divide and undergo apoptosis throughout life. However, the normal adult artery shows very low apoptotic and mitotic indices. In diseased tissue additional factors are present both locally, such as inflammatory cytokines, inflammatory cells, and the presence of modified cholesterol, and systemically, such as blood pressure and flow. These factors substantially alter the normal balance of proliferation and apoptosis, and apoptosis in particular may predominate in many disease states.
Remodelling
Remodelling defines a condition in which alterations in vessel size can occur through processes that do not necessarily require large changes in overall cell number or tissue mass. For example, physiological remodelling by cell proliferation/apoptosis results in closure of the ductus arteriosus and reduction in lumen size of infra-umbilical arteries after birth, and remodelling occurs in primary atherosclerosis, after angioplasty and in restenosis. Although surgical reduction in flow results in compensatory VSMC apoptosis, the role of VSMC apoptosis per se in determining the outcome of remodelling is unclear.
Arterial injury and aneurysm formation
Acute arterial injury at angioplasty is followed by rapid induction of medial cell apoptosis. In animal models injury results in medial cell apoptosis 30 minutes to six hours after injury8 with adventitial and neointimal apoptosis occurring later. In humans, restenosis after angioplasty has been reported to be associated with either an increase or decrease in VSMC apoptosis, and again the role of VSMC apoptosis in either the initial injury or the remodelling process in restenosis in humans requires further study.
The most common form of arterial aneurysm in humans is characterised by a loss of VSMCs from the vessel media, with fragmentation of elastin and matrix degradation, leading to progressive dilatation and eventually rupture. Apoptosis of VSMCs is increased in aortic aneurysms compared with normal aorta, associated with an increase in expression of a number of pro-apoptotic molecules. In particular, the presence of macrophages and T lymphocytes in aneurysms suggests that inflammatory mediators released by these cells may promote VSMC apoptosis. Moreover, the production of tissue metalloproteinases by macrophages may accelerate apoptosis by degrading the extracellular matrix from which VSMCs derive survival signals (see below).
Atherosclerosis
Rupture of atherosclerotic plaques is associated with a thinning of the VSMC-rich fibrous cap overlying the core. Rupture occurs particularly at the plaque shoulders, which exhibit lack of VSMCs and the presence of inflammatory cells. Apoptotic VSMCs are evident in advanced human plaques including the shoulder regions, prompting the suggestion that VSMC apoptosis may hasten plaque rupture. Indeed, increased VSMC apoptosis occurs in unstable versus stable angina lesions.
Although loss of VSMCs would be expected to promote plaque rupture, there is no direct evidence of the effect of apoptosis per se in advanced human atherosclerosis. Most apoptotic cells in advanced lesions are macrophages next to the lipid core.9 Loss of macrophages from atherosclerotic lesions would be predicted to promote plaque stability rather than rupture, since macrophages can promote VSMC apoptosis by both direct interactions and by release of cytokines. However, macrophage apoptosis is found at sites of plaque rupture,10 although it is not known if death directly promotes rupture, or simply that macrophages are the most common cell types found at rupture sites.
Effect of VSMC apoptosis
The effect of VSMC apoptosis is clearly context dependent. Thus, intimal VSMC apoptosis in advanced atherosclerotic plaques may promote plaque rupture, or medial apoptosis may promote aneurysm formation. In neointima formation post-injury, VSMC apoptosis of both intima and media can limit neointimal formation at a defined time point. However, apoptosis is also associated with a number of deleterious effects. Exposure of phosphatidylserine on the surface of apoptotic cells provides a potent substrate for the generation of thrombin and activation of the coagulation cascade,11 and apoptotic cells release membrane bound microparticles that are systemically procoagulant. Finally, VSMC apoptosis may be directly pro-inflammatory, with release of chemoattractants and cytokines from inflammatory cells.
REGULATION OF APOPTOSIS
Apoptosis via death receptors
Many stimuli can trigger apoptosis, but in vascular disease specific alterations within the cell elicit sensitivity to a particular stimulus that is disease associated. Thus, remodelling may trigger apoptosis following reduction in blood flow, the major stimulus being flow dependent stimuli such as nitric oxide or shear stress. In contrast, VSMC apoptosis in atherosclerosis or aneurysm formation may be caused by inflammatory cells that express surface death ligands or secrete pro-apoptotic cytokines. Whatever the stimulus, most downstream pathways that signal apoptosis are similar.
Diseases in which apoptosis has been implicated
-
▸ Cardiac (myocyte)
-
idiopathic dilated cardiomyopathy
-
ischaemic cardiomyopathy
-
acute myocardial infarction
-
arrhythmogenic right ventricular dysplasia
-
myocarditis
-
-
▸ Cardiac (conducting tissues)
-
pre-excitation syndromes
-
heart block, congenital complete atrioventricular heart block, long QT syndromes
-
-
▸ Vascular
-
atherosclerosis
-
restenosis after angioplasty/stenting
-
vascular graft rejection
-
arterial aneurysm formation
-
The regulation of apoptosis can be simplified into two major pathways (figs 2 and 3). First, membrane bound death receptors of the tumour necrosis receptor family (TNF-R), such as Fas (CD95), TNF-R1, or death receptors (DR) 3–6, bind their trimerised ligands causing receptor aggregation, and subsequent recruitment of adapter proteins (Fas-FADD, TNF-R1-TRADD, etc) through protein:protein interactions12, 13 (fig 2). In turn, adapters recruit cysteine proteases (caspases) such as caspase 8 (FLICE) and caspase 2 to the complex.14 Within the complex of Fas, FADD, and caspase 8 (known as the death inducing signalling complex (DISC)), caspase 8 becomes proteolytically activated by oligomerisation.15 This in turn activates the terminal effector caspases (caspases 3, 6, and 7) responsible for cleavage of intracellular substrates required for cellular survival, architecture, and metabolic function.

Schematic of Fas death signalling pathways. Fas, the prototypic member of the tumour necrosis factor (TNF) death receptor family, binds to its cognate ligand. Recruitment of the adapter molecule FADD and pro-caspase 8 results in activation of the latter. Caspase 8 activation directly activates downstream caspases, (3, 6, and 7) which results in DNA fragmentation and cleavage of cellular proteins. This pathway is thought to occur in type I cells and does not involve mitochondrial pathways. Caspase 8 activation also results in cleavage of Bid, which translocates and interacts with other Bcl-2 family members (see fig 3).

{kind=link}
{kind=link}
{kind=link}
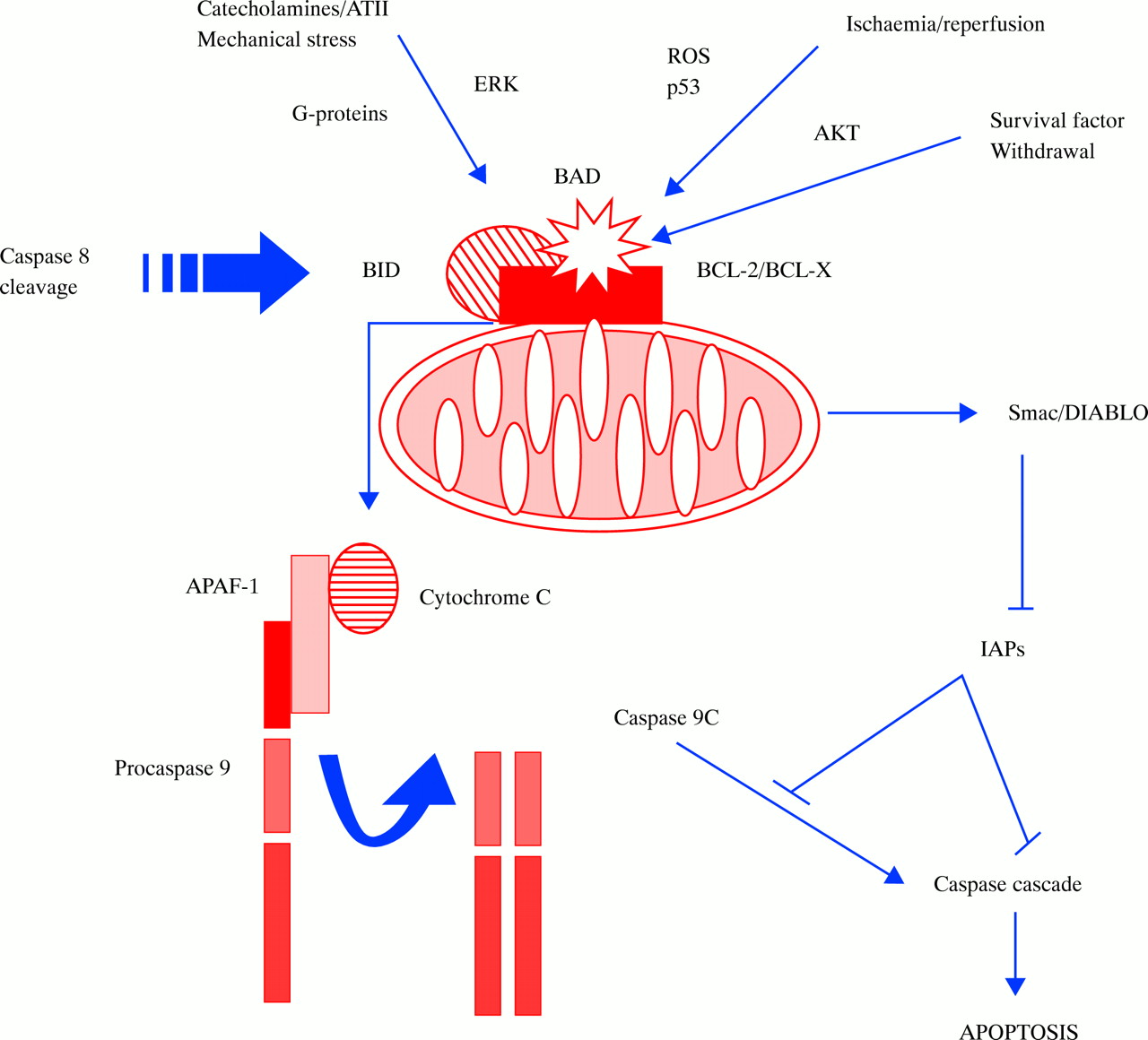
Schematic of mitochondrial death signalling pathways. Anti-apoptotic members of the Bcl-2 family, such as Bcl-2 and Bcl-X, are located on the mitochondrial outer membrane. Here they act to prevent the release of apoptogenic factors from the inner mitochondrial space. Binding of the pro-apoptotic proteins Bid (after cleavage by caspase 8) or Bad (after dephosphorylation) to Bcl-2 mitigates the protective effect of Bcl-2 and triggers release of cytochrome c and Smac/DIABLO. Cytochrome c, in concert with the adapter protein apaf-1 and caspase 9, activates caspase 3 and the downstream caspase cascade. Smac/DIABLO inhibits IAPs (inhibitor of apoptosis proteins), which in turn inhibit caspase activities, thus propagating apoptosis. Stimuli such as growth factor withdrawal or activation of p53 and Fas activation in type II cells act through this mitochondrial pathway.
Apoptosis via mitochondrial amplification
In addition to direct activation of caspases, caspase 8 activation causes cleavage of bcl-2 family proteins such as bid (fig 3). Bcl-2 family members are either pro-apoptotic (Bax, Bid, Bik, Bak) or anti-apoptotic (Bcl-2, Bcl-XL). Activation of pro-apoptotic Bcl-2 family members causes their translocation to mitochondria, where they interact with anti-apoptotic members that are mitochondrial membrane components. This interaction depolarises voltage dependent mitochondrial channels and releases mitochondrial mediators of apoptosis such as cytochrome c16 and Smac/DIABLO. The association of cytochrome c with an adapter molecule apaf-1 and caspase 9 activates caspase 3, and the caspase cascade. In contrast, Smac/DIABLO promotes apoptosis by directly antagonising inhibitor of apoptosis proteins (IAPs) (see below).
Apoptosis can also be blocked by expression of several intracellular proteins, including FLIPs (FLICE inhibitory proteins) and IAPs (fig 2). FLIPs have the same pro-domain structure as caspase 8, but do not the active caspase site within the C-terminus. Binding of FLIP to caspase 8 therefore prevents its activation. In contrast, IAPs inhibit the enzymatic activity of downstream caspases, or they can mediate anti-apoptotic signalling pathways through the activation of nuclear transcription factor κβ.
REGULATION OF CARDIOMYOCYTE APOPTOSIS
The stimulus for cardiomyocyte apoptosis clearly depends upon the clinical or experimental setting. Ischaemia is associated with many changes in the intracellular and extracellular milieu of cardiomyocytes, many of which are potent apoptotic stimuli. Thus, hypoxia promotes cardiomyocyte apoptosis, both in vitro and in vivo, and ischaemia/reperfusion and hypoxia/reoxygenation are associated with increased expression of Fas. Decreased serum and glucose concentrations trigger cytochrome c release from mitochondria in cardiomyocytes, suggesting that ischaemia induced apoptosis may be mediated by mitochondrial amplification. Indeed oxygen species promote apoptosis by triggering pathways involving mitochondrial release of cytochrome c and caspase activation.
In heart failure, a huge variety of initial stimuli have been propounded. In vitro, mechanical stretch can induce apoptosis, indicating a possible role for volume overload and raised ventricular end diastolic pressure; pressure overload following aortic banding also induces early myocyte apoptosis, before significant hypertrophy. Both four weeks of rapid ventricular pacing and catecholamines induce myocytes apoptosis in dogs associated with heart failure, suggesting that catecholamine responses may be directly toxic to myocytes.
REGULATION OF VASCULAR SMOOTH MUSCLE CELL APOPTOSIS
Human VSMCs express death receptors, and inflammatory cells within the atherosclerotic plaque express death ligands; interaction between membrane bound ligands and receptors may therefore induce VSMC death. In contrast, soluble ligand binding to death receptors is a very weak inducer of VSMC apoptosis, and does not induce apoptosis in the absence of “priming” of the cell. Some of this resistance can be explained by intracellular location of death receptors in VSMCs,17 and priming may be associated with increased receptor expression. Physiologically, combinations of cytokines such as interleukin (IL) β (IL-1β), interferon γ (IFNγ) and tumour necrosis factor α (TNFα) increase surface death receptors, possibly via nitric oxide and p53 stabilisation.
Irrespective of the local environment, VSMCs derived from atherosclerotic plaques are intrinsically sensitive to apoptosis,18 compared with cells from normal vessels. Heterogeneity of sensitivity between VSMCs in the vessel wall is also seen in animal vessels after injury, and in medial VSMCs from normal human arteries, This reflects differences in expression of pro- and anti-apoptotic molecules, specifically those regulating signalling from survival cytokines, cell:cell and cell:matrix interactions, and members of the bcl-2 family. This may underlie observations that despite (apparently) the same stimulus for apoptosis, VSMC apoptosis in either normal or diseased vessels wall is highly localised. Indeed, insulin-like growth factor 1 receptor concentrations (IGF-1R), a potent survival signalling system for normal VSMCs, are downregulated in plaque VSMCs.
The bcl-2 family members are critical in regulating VSMC apoptosis, both in vitro and in vivo. Human VSMCs express low levels of Bcl-2, but Bax is expressed in atherosclerotic plaques; reduced levels of VSMC apoptosis seen after cholesterol lowering in rabbit models of atherosclerosis is accompanied by a loss of Bax immunoreactivity. In vivo, rat VSMCs express minimal Bcl-2, but high levels of Bcl-X can be found after injury. Indeed, inhibition of Bcl-X dramatically induces apoptosis of VSMCs after balloon injury19 and differences in expression of Bcl-X may account for differences in apoptosis sensitivity of intimal versus medial VSMCs. Regulation of sensitivity to apoptosis in VSMCs is also mediated by expression of IAP proteins and individual caspases.
THERAPEUTIC OPTIONS FOR APOPTOSIS TREATMENT
The prevention of cardiomyocyte apoptosis is now a very important therapeutic aim. However, critical to determining therapeutic benefit is not just inhibiting apoptosis markers at a single defined time point, but actually improving cardiac function. Many agents prevent the development of the morphological appearance of apoptosis or a biochemical marker (for example, DNA fragmentation) without inhibiting cell death. The ability to delay death may serve no useful purpose and may even be deleterious if that cell undergoes subsequent necrosis, with concomitant inflammation. In contrast, some studies have indicated that inhibition of apoptosis improves ventricular remodelling and contractility after infarction.20 Although the long term effects of this inhibition are unknown, clinically meaningful improvements in cardiac function have been achieved.
Apoptosis can be interrupted at many points in the signalling pathway. Prevention of apoptotic myocyte death may be directed at (1) inhibiting/preventing the stimulus, (2) inhibiting the regulatory mechanisms determining the decision to die, or (3) inhibiting the pathways executing apoptosis. The cascade of events leading to cardiomyocyte apoptosis, and also the point at which a cell is irreversibly committed to die, crucially determine the approach to inhibiting apoptosis. Clearly, many signalling pathways are activated in ischaemia and heart failure. Interruption of a single pathway may therefore not inhibit apoptosis if there are multiple, redundant pathways inducing apoptosis.
In contrast, mediators that act beyond convergence of multiple signalling pathways may be better targets to inhibit apoptosis. However, many of the identified downstream mediators are enzymes required for effective cell disintegration and packaging, and may be beyond the point at which the cell is committed to die. Inhibition here would prevent the cellular appearances and markers of apoptosis, but the cell would still die. In addition, these molecules are critical to apoptosis in many tissues and such non-cardiac specificity may be unwelcome. From this argument, inhibiting the stimulus to apoptosis, particularly if specific to the heart at one point in time, would be more effective. The timing and delivery of treatment is also dependent upon the clinical situation. Clearly, it is easier to inhibit apoptosis transiently in an acute situation, such as myocardial infarction, than with chronic treatment in heart failure.
Inhibiting/preventing the pro-apoptotic stimulus
Ischaemia/reperfusion, hypertrophy caused by increased afterload, and myocardial remodelling following infarction all are associated with myocyte apoptosis. This suggests that current treatment of proven benefit in these diseases may already act by inhibition of apoptosis. The beneficial effects of β blockers in chronic heart failure and ischaemic heart disease may counteract the pro-apoptotic effect of excess catecholamines. Indeed, carvedilol can inhibit ischaemia/reperfusion induced myocyte apoptosis, and angiotensin converting enzyme inhibitors may protect against angiotensin II induced apoptosis. Clearly, approaches aimed at reducing myocardial stretch, or oxidative stress, or improving myocardial perfusion may have the same effect. Finally, many pathways leading to apoptosis are triggered by specific death ligands, with either apoptosis or the disease itself manifesting upregulation of death receptors. Inhibition of delivery of death ligands—for example, by scavenging ligands through soluble receptors or receptor antagonists—may reduce apoptosis mediated though these pathways. However, it should be noted that other signals emanate from death receptors. For example, Fas activation reduces the membrane potential and induces afterdepolarisations in cardiac myocytes; inhibiting Fas induced apoptosis may allow escape of other Fas signalling, promoting arrhythmias.
Protection against apoptosis
Many molecules protect cells from apoptosis, including anti-apoptotic Bcl-2 family members, IAPs, and decoys for death receptors. Although these agents inhibit apoptosis mediated by many stimuli, and may therefore be clinically useful, at present they cannot be selectively expressed without gene transfer into the heart, with all its inherent problems. More promising is the potential administration of soluble survival factors following the apoptotic stimulus. Many growth factors, including IGF-1, cardiotrophin-1, and the neuregulins, inhibit apoptosis following ischaemia, serum withdrawal, myocyte stretch, and cytotoxic drugs. Indeed, overexpression of IGF-1 reduces apoptosis in non-infarcted remote zones and promotes favourable remodelling postmyocardial infarction.20 Activation of the cardiotrophin-1 receptor also inhibits cardiac dilatation following aortic banding, suggesting that reduced cardiomyocyte apoptosis can be translated into improved function. These agents signal through the AKT and ERK pathways, respectively, that are known to be anti-apoptotic in many cell types (table 2).
Potential inhibitors and signalling pathways of cardiomyocyte apoptosis
Apoptosis in the cardiovascular system: key points
-
▸ Apoptosis of cardiomyocytes is seen in acute myocardial infarction where it may contribute to infarct size, and also in chronic heart failure, where it may be responsible for the gradual decline in cardiac function
-
▸ Apoptosis of vascular smooth muscle cells is both physiological, in vessel remodelling, and pathological, in disease states such as atherosclerosis and arterial aneurysm formation
-
▸ Apoptosis is regulated by both pro- and anti-apoptotic stimuli, and both activators and inhibitors of apoptosis signalling within the cell
-
▸ Treatment to inhibit apoptosis in the heart can be targeted to inhibit ischaemia or reperfusion injury, to enhance endogenous protective mechanisms within cardiomyocytes, or to disrupt apoptosis signalling
-
▸ The benefits of conventional heart failure treatment may be due in part to the inhibition of cardiomyocyte apoptosis
In contrast, some agents are potential therapeutics for long term administration. Heart failure is characterised by increased plasma concentrations of catecholamines and TNFα. The beneficial effects of β blockers in heart failure may therefore be achieved by prevention of myocyte apoptosis. Licensed inhibitors of TNFα are now available, although recent randomised controlled trials (RENAISSANCE and RECOVER) suggest that a soluble TNF receptor antagonist (etanercept) does not benefit patients with heart failure. In contrast, evidence identifying the type 2 angiotensin II receptor as inducing apoptosis in models of heart failure has suggested that its inhibition may be beneficial.
Preventing execution of apoptosis
Execution of apoptosis and cellular disintegration and packaging requires the activation of downstream signalling pathways, including mitochondrial amplification and activation of caspases. Augmentation of endogenous inhibitors of caspases, such as the IAPs, could therefore inhibit apoptosis induced by many stimuli. Pharmacological inhibition of caspases using cell permeable analogues of cleavage sites can inhibit myocyte apoptosis over the short term. However, their long term benefits are unknown, as cells that are destined to die may do so anyway, and delaying apoptosis may not provide long term benefit.
CONCLUSION
VSMC apoptosis occurs in the vasculature in both physiological and pathological contexts. Deaths are regulated by specific proteins that serve either to induce or protect against apoptosis. We are now beginning to understand the complex pro- and anti-apoptotic factors that lead to cell loss from the vasculature. Sensitivity to apoptosis is determined by expression of cell death receptors and ligands, and by multiple protein species below receptor level. In addition, sensitivity is determined by the presence and response to survival cytokines, mitogens, and local cell and matrix interactions, and by the growth status of the cell. Although much research has been performed in vitro, future studies in vivo should identify which pro- and anti-apoptotic factors are functional in vivo.
Apoptosis of cardiac myocytes is part of many disease states, including myocardial infarction and heart failure. At present, the precise role of cardiomyocyte apoptosis in the pathogenesis of these diseases is unknown, and therefore the benefit from anti-apoptotic treatment is unproven. Prevention of cardiomyocyte apoptosis may involve inhibiting both the pro-apoptotic stimulus and apoptosis signalling within the cell. Given the lack of cardiac specificity of apoptosis signalling, such strategies may benefit short lived insults, such as myocardial infarction or unstable angina, rather than heart failure. However, it is also highly likely that proven conventional treatment for heart failure works at least in part by inhibiting apoptosis.
REFERENCES
Linked Articles
- Miscellanea
- Miscellanea