Article Text

Statistics from Altmetric.com
- Bicuspid aortic valve
- thoracic aortic aneurysm
- aortic dissection
- aortic disease
- valvular heart disease
- dissection
- aortic valve disease
Bicuspid aortic valve (BAV) is one of the most common congenital heart defects. This condition is associated with significant valvular disease including aortic stenosis, aortic regurgitation, and infective endocarditis. BAV disease not only affects the aortic valve, but also the aortic root, ascending aorta, and aortic arch, all tissues originating from the neural crest. BAV may be associated with important diseases of the aorta including ascending aortic dilatation, ascending aortic aneurysm, and coarctation of the aorta. Notably, BAV is a well recognised risk factor for acute aortic dissection. It may occur sporadically or can occur as a familial trait. Additional congenital heart defects may be associated with BAV.
With enhanced imaging and discoveries in molecular genetics and cellular research, the aortopathy of BAV disease is becoming much better understood. Underlying alterations in connective tissue, metalloproteinase and inhibitor activity, cell signalling, and specific gene mutations are all being evaluated in BAV aortic disease. This article discusses the key points in the pathophysiology, evaluation, and management of the aortic involvement in patients with BAV.
BAV is common
The prevalence of BAV is between 0.5–1.4% of the general population and has a male predominance of at least 2–3:1 in most series.1 There is a wide spectrum of BAVs with variability in appearance and morphology. Most often (∼75%), the right and left coronary leaflets are the larger, fused leaflet, with the leaflets oriented right and left and the true commissures oriented in an anterior and posterior manner (figure 1). Certain populations have a much higher prevalence of BAV, including those with coarctation of the aorta and Turner syndrome. Because of the high prevalence of BAV in the general population, BAV is one of the most common underlying disorders requiring aortic valve replacement. Additionally, because it is a common disorder and many patients with BAV have coexisting aortic root and ascending aortic dilatation, BAV is one of the conditions most commonly associated with a dilated ascending aorta. One series found that 20% of patients discovered to have a dilated aortic root on echocardiogram had a coexistent BAV.1 In our clinic, BAV is by far the most common underlying disorder for which a dilated aortic root is being investigated. If the aortic valve morphology is not clearly delineated in the patient with unexplained dilatation of the ascending aorta, I often recommend a transoesophageal echocardiogram (or MRI) to evaluate for a BAV.

Transthoracic echocardiogram of bicuspid aortic valve (BAV). (A) Short axis view of the BAV in diastole demonstrating asymmetrical sinuses and a raphe. (B) BAV in systole with elliptical opening pattern. Reproduced with permission from Braverman and Beardslee.1
BAV may be associated with other cardiovascular lesions
While BAV may occur in isolation, its presence should prompt a full evaluation for associated cardiovascular and congenital defects. Coarctation of the aorta (CoA), Turner syndrome, sinus of Valsalva aneurysm, ascending aortic aneurysm, aortic dissection, and many congenital heart defects are all associated with BAV (box 1, figures 2 and 3).
Cardiovascular conditions associated with bicuspid aortic valve
Coarctation of the aorta
Turner syndrome
Coronary artery anomalies
Sinus of Valsalva aneurysm
Ascending aortic aneurysm
Aortic dissection
Supravalvular aortic stenosis
Patent ductus arteriosus
Ventricular septal defect
Shone's complex*
Familial thoracic aortic aneurysm/dissection syndromes
*Shone's complex is a congenital anomaly which consists of a series of left heart obstructive lesions including supravalvular mitral ring, parachute mitral valve, subaortic stenosis, and coarctation of the aorta. A bicuspid aortic valve is noted in ∼70% of patients.

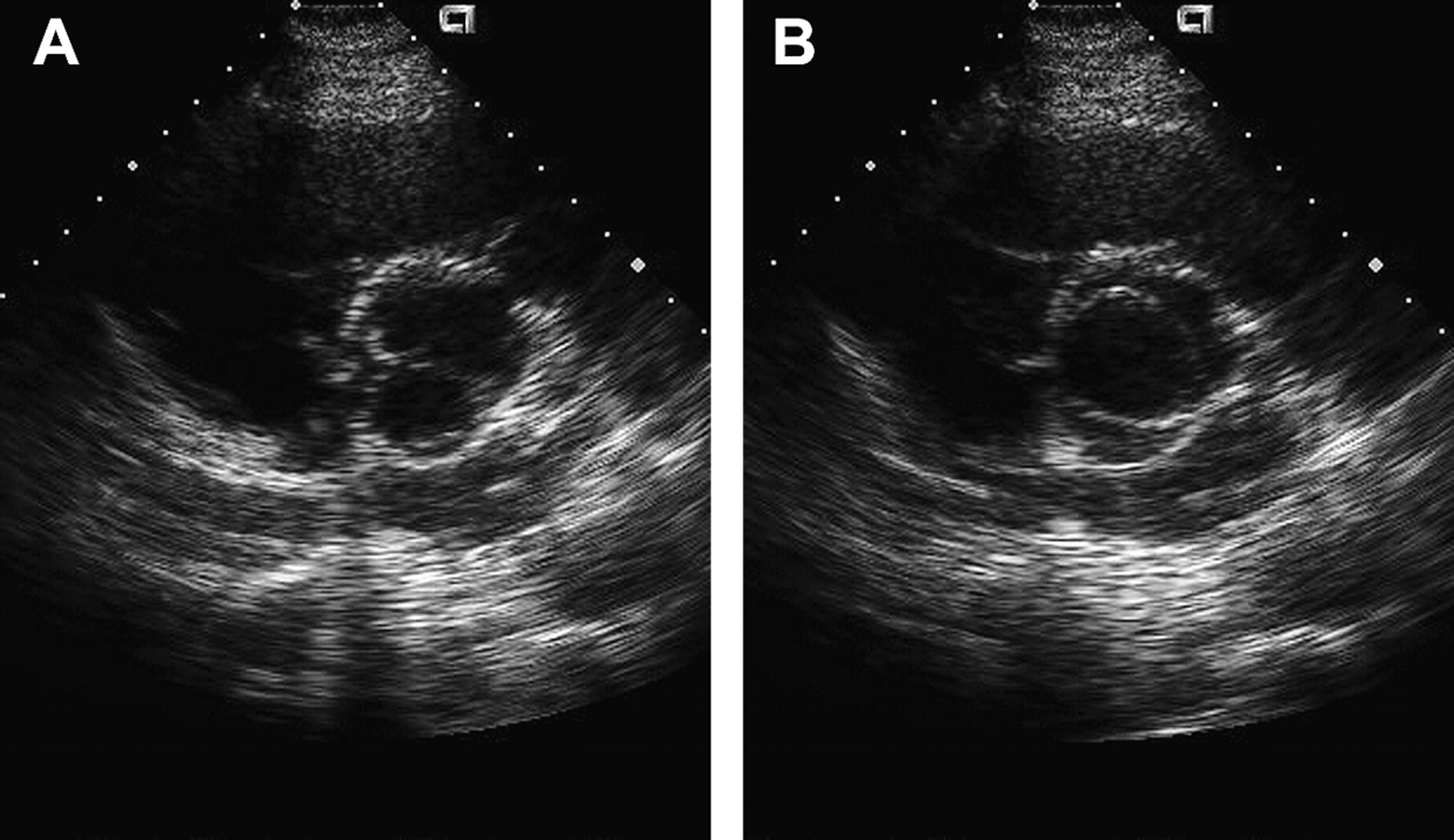
Transoesophageal echocardiogram of a bicuspid aortic valve (BAV) and sinus of Valsalva aneurysm. (A) Transoesophageal echocardiogram long axis view demonstrating aneurysm of the non-coronary cusp. (B) Short axis image demonstrating BAV and dilated aortic sinus. Reproduced with permission from Braverman and Beardslee.1

Representative cardiovascular MRI of a patient with a coarctation of the aorta. Note the typical postductal stenosis (arrow). Reproduced with permission from Braverman AC, Guven H, Beardslee MA, et al. The bicuspid aortic valve. Current Prob Cardiol 2005;30:470–522.
The natural history of BAV associated with CoA led to initial theories about an underlying aortic medial defect associated with the condition.1 Over 80 years ago, Abbott recognised the increased risk of aortic dissection in the setting of a CoA.1 However, when a BAV coexisted with CoA, the risk of aortic dissection was twice as high as the risk of dissection with CoA alone.1 In a more recent series of patients with CoA, the presence of a BAV was the most powerful clinical predictor of complications involving the aorta—including aortic aneurysm, aortic dissection, and rupture.2
Turner syndrome, affecting 1 in 3000 newborn females, is characterised by complete or partial absence of one X chromosome. Clinical features include short stature, webbing of the neck, low hairline, and a shield-like chest. Cardiovascular defects are noted in up to 75% of patients with Turner syndrome, including BAV, CoA, pseudo-CoA, and partial anomalous pulmonary venous return. Turner syndrome is complicated by BAV in up to 30% of cases and most are due to fusion of the right and left coronary cusps. Patients with Turner syndrome, BAV, and aortic root dilatation are at increased risk for aortic dissection. Because of their small body size, patients with Turner syndrome should undergo elective ascending aortic aneurysm replacement at a much smaller absolute size and the aortic size should be indexed to body surface area.3
BAV is associated with aortic dissection
BAV is a well recognised risk factor for aortic dissection, even in the absence of hypertension, CoA, aortic stenosis or regurgitation (figure 4).1 4 In autopsy reports of patients with aortic dissection, a BAV is reported in 7–9% of patients. In clinical series of aortic dissection, BAV is present in 4–7% of individuals and in up to 15% of those suffering ascending aortic dissection. When one examines aortic dissection among the young (ie, <40 years old), BAV is reported in 9–28% of cases.1 In the International Registry of Acute Aortic Dissection (IRAD), a BAV was present in 9% of young patients with aortic dissection, but only in 1% of those >40 years old.5 From these data and other series, it has been stated that aortic dissection occurs 5–10 times more commonly among patients with BAVs compared to those with trileaflet aortic valves (TAVs).1 Risk factors for aortic dissection in the setting of BAV have been reported and include absolute aortic diameter, male gender, family history, coarctation of the aorta, and Turner syndrome. Additionally, aortic dissection among those with a BAV occurs on average one decade earlier than aortic dissection in the setting of a TAV.1 However, other series have not reported high frequencies of aortic dissection in BAV patients. In patients with known BAV followed prospectively, the risk of aortic dissection has been an uncommon event.6 7
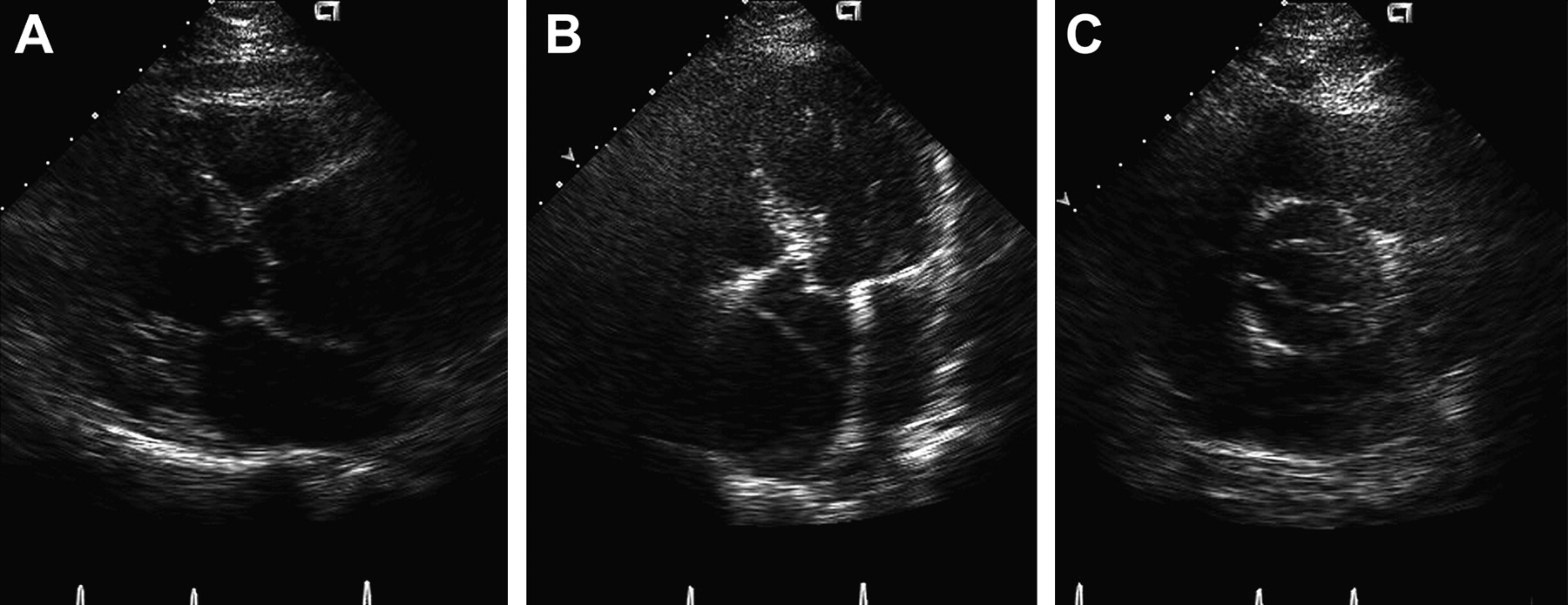
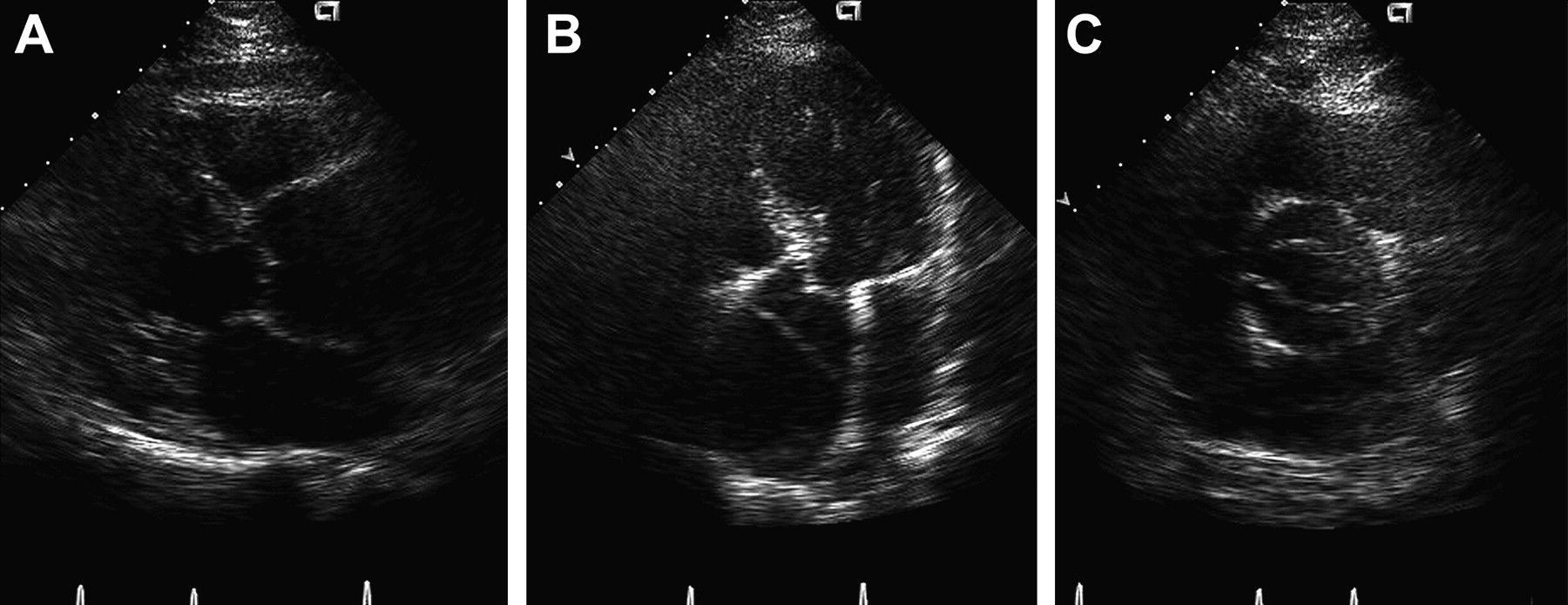
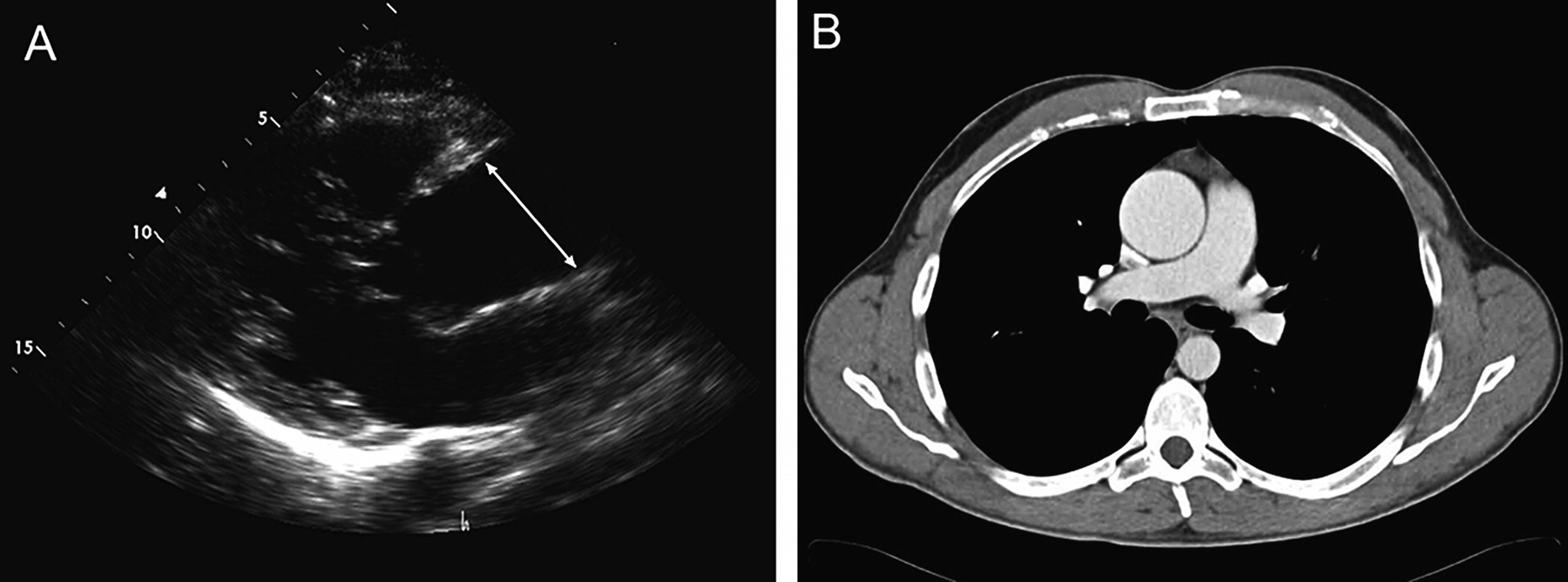
Transthoracic echocardiogram of a bicuspid aortic valve with severe aortic root dilatation and aortic dissection. (A) Parasternal long axis view with severe aortic root dilatation. (B) Apical five chamber view demonstrating the intimal flap and a dilated aortic root. (C) Short axis view demonstrating the bicuspid aortic valve. Reproduced with permission from Braverman AC, Guven H, Beardslee MA, et al. The bicuspid aortic valve. Curr Prob Cardiol 2005;30:470–522.
Ascending aortic dilatation and aneurysm occur commonly with BAV
In years past, the dilated aorta associated with underlying aortic valve disease was ascribed to ‘post-stenotic’ dilatation. It has been recognised that the BAV demonstrates abnormal leaflet motion, including folding or wrinkling of the valve tissue and increased leaflet doming during the cardiac cycle. This may result in increased currents of turbulence even when the leaflets are not stenotic.1 Because aortic dilatation is found in the setting of BAV, even when the valve has no associated aortic stenosis or regurgitation and late after valve replacement, the haemodynamic alterations are not considered to be the primary underlying mechanism. Instead, underlying abnormalities of the aortic media are considered the primary aetiology for the aortic dilatation associated with BAV.
Echocardiogram surveys of BAV patients have demonstrated that patients with BAVs have larger aortic dimensions at the annulus, sinuses of Valsalva, and ascending aorta compared to matched patients with TAVs (figure 5).1 8 However, when underlying valve dysfunction is present, the type of valve lesion does correlate with the degree of aortic enlargement, with greater aortic dimensions present when there is more severe aortic regurgitation.1 9 Thus, haemodynamic stresses related to increased stroke volume, turbulence, aortic curvature, and other haemodynamic factors may play a facilitative role in the aortic dilatation and complications of BAV.

Aortic root aneurysm complicating a functionally normal bicuspid aortic valve. (A) Transthoracic echocardiogram of a 4.9 cm ascending aorta. (B) Corresponding CT image of the aortic root aneurysm. Reproduced with permission from Braverman and Beardslee.1
Aortic root and ascending aortic dilatation have been recognised in patients with BAV, whether the aortic valve function is ‘normal’ or when there is associated aortic stenosis or regurgitation, and this may be recognised early in life.1 10 Echocardiographic surveys in children with BAV have demonstrated a larger aortic root and ascending aorta compared to children with TAV, and this enlargement is independent of any aortic valve stenosis or regurgitation.1 Not all patients with BAV have aortic dilatation and the prevalence varies depending upon the study and criteria selected.
Non-invasive methods to evaluate aortic wall biomechanical properties have demonstrated reduced aortic elasticity and reduced aortic distensibility in patients with BAV. Additionally, leaflet orientation of the BAV has been reported to correlate with aortic elastic properties.1
The aortic enlargement associated with BAV is progressive with aortic dilatation being reported in ∼50% of those <30 years to up to 90% in those >80 years, depending upon the specific definitions used for aortic dilatation.9 In a study from Olmsted County, Minnesota, the prevalence of aortic root dilatation >40 mm was 15%, and in a group of patients in whom there was repeated measurements over time, the prevalence of aortic dilatation increased to nearly 40%.6 There is variable information about the rate of progression of aortic disease associated with a BAV. In a study of children with BAV, the mean rate of ascending aortic growth was 1.2 mm/year.1 Series in adults have reported ascending aortic growth rates of between 0.2–0.19 mm/year, while aortas which are already aneurysmal demonstrate more rapid growth.1 Some studies have reported that, compared to aneurysms associated with a TAV, those with a BAV demonstrate more rapid growth.1
Aortic root and ascending aorta must be visualised in BAV disease
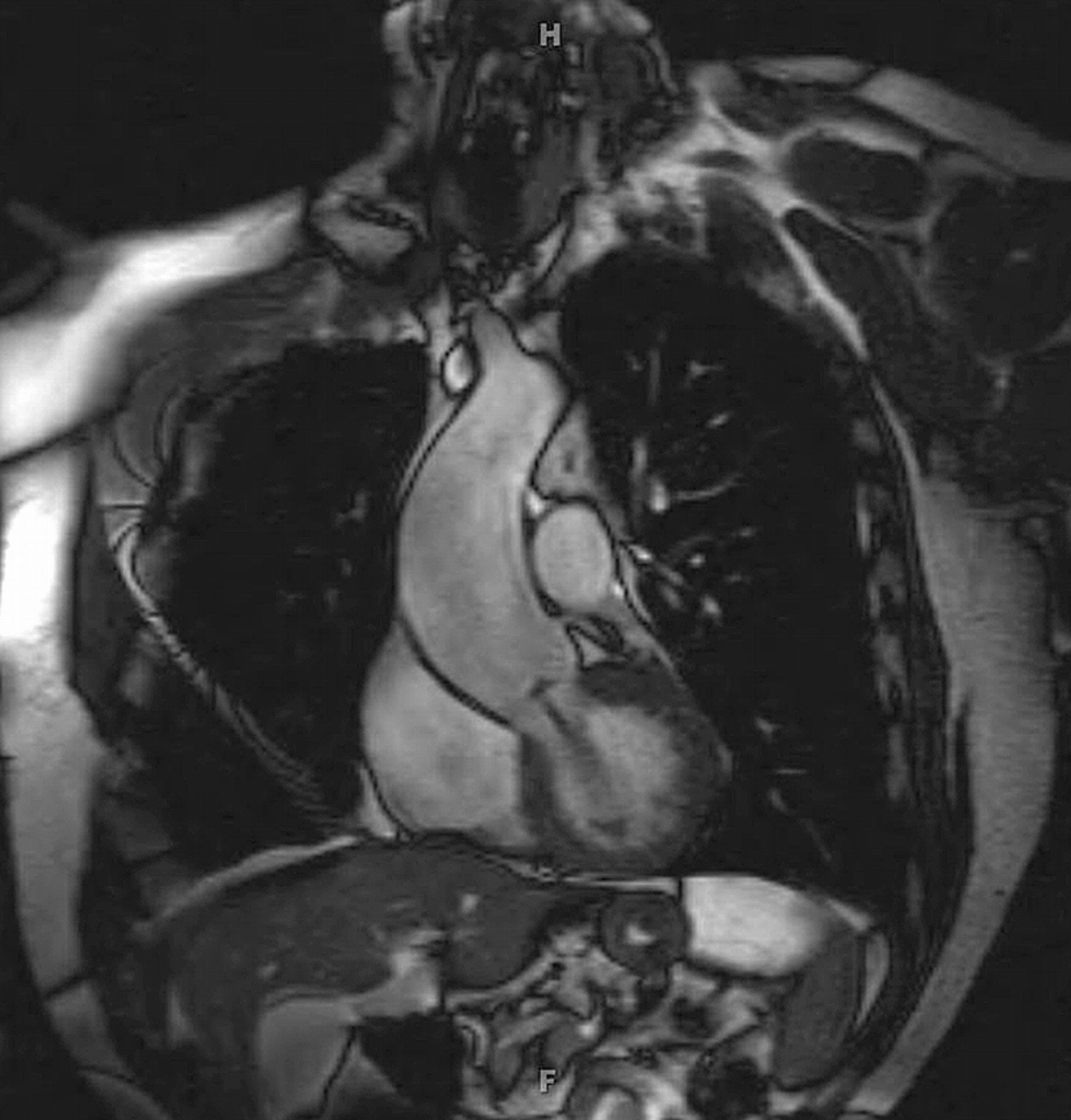
While aortic dilatation may begin in the sinuses of Valsalva, the typical patient with BAV and aortic dilatation has enlargement most pronounced in the proximal to mid ascending aorta (figure 5). This is very important to recognise, as the standard transthoracic echocardiogram may not visualise the entire ascending aorta and may fail to notice the largest diameter of the aorta in the setting of BAV disease. It is recommended that an MRI or CT scan be performed to evaluate the ascending aorta in the setting of BAV disease when the entirety of the ascending aortic dilatation is not adequately visualised by echocardiogram (figure 6). In the setting of aortic dilatation or aneurysmal enlargement, I recommend a CT or MRI scan to evaluate the thoracic aorta fully. It has been reported that distinct aortic root phenotypes may be associated with various leaflet orientations in BAV, but others have not demonstrated a specific relationship between BAV cusp orientation and the degree or type of aortic dilatation.

Cardiac MRI of a dilated ascending aorta associated with a bicuspid aortic valve.
BAV is associated with cystic medial degeneration
The histopathologic abnormality underlying the ascending aortic complications in BAV is cystic medial degeneration (figure 7). This pathologic finding has been demonstrated in the aortas of patients with BAV before aneurysm formation occurs.1 Apoptosis may be one of the mechanisms responsible for aortic medial smooth muscle cell loss associated with aortic dilatation and aneurysm formation in the setting of BAV disease.1 11 Focal apoptosis of significant degree has been demonstrated in the aortic wall in patients with BAV, even in the absence of aneurysm formation.1 11 Interestingly, when aortic aneurysm complicates a BAV, the degree and pattern of medial degeneration differs from aneurysms associated with a TAV. BAV aneurysms have increased apoptosis, greater degrees of elastic fragmentation, and alterations in tissue concentrations of matrix metalloproteinase (MMP) and endogenous inhibitors.12 Elevation in MMP-2 expression is found in BAV aortic aneurysms, while the level of MMP-9 activity is normal. Expression of regulators of MMP activity, such as protein kinase C, differs in BAV aneurysms compared to those aneurysms associated with TAVs. Thus, the cystic medial degeneration associated with BAV disease may be related to imbalance between proteolytic enzymes and their inhibitors in the aortic wall. Plasma concentrations of MMP-2 have also been reported to be increased in BAV patients with dilated aortas compared to BAV patients without aortic dilatation and normal controls.7

Elastic tissue stain of aortic media from a normal aorta from a trileaflet aortic valve patient demonstrating normal elastic fibre orientation (labelled Normal Aorta); and elastic stain from a patient with a bicuspid aortic valve and aortic root aneurysm demonstrating cystic medial degeneration (labelled Aortic Aneurysm). Courtesy of Robert Thompson, MD. Reproduced with permission from Braverman AC, Guven H, Beardslee MA, et al. The bicuspid aortic valve. Curr Prob Cardiol 2005;30:470–522.
In Marfan syndrome, FBN1 gene mutations lead to altered structure and function of the matrix protein fibrillin-1, which in turn leads to a cascade of events including altered TGF-β signalling.4 Lower content of fibrillin-1 has been reported in BAV aortas compared to those with TAVs, but it is not known whether this finding is a primary or secondary event.1 4 A similar degree of apoptosis and alterations in matrix proteins have been reported in vascular smooth muscle cells from those with BAV aneurysms and those with Marfan syndrome.13 Altered TGF-β signalling has been hypothesised to play a potential role in BAV aneurysm disease, but this is unproven.1
BAV and BAV aortic aneurysm may be familial
BAVs may occur in families with a much higher prevalence than is recognised in the general population. In large family studies, the prevalence of BAVs in first degree relatives of an individual with a BAV has been reported to be 9%.14 The inheritance of BAV is consistent with an autosomal dominant pattern with variable expressivity and reduced penetrance. Animal models of BAV disease have reported abnormalities in endothelial nitric oxide synthase, NKX2.5, and NOTCH signalling. NOTCH1 mutations have also been found in a small number of families with BAV.
Importantly, BAV associated with ascending aortic aneurysm may also be familial. Potential loci for BAVs and ascending aortic aneurysms have been suggested at multiple sites, including 15q, 18q, 5q, 13q, and NOTCH1.1 Loscalzo et al reported on many families with BAV and ascending aortic aneurysms and noted that many family members had thoracic aortic aneurysm or aortic dissection whether or not a BAV was present.15 Abnormal aortic elastic properties have been demonstrated in the relatives of individuals with BAV disease, even in the absence of aortic dilatation.16 Thus, it is important to screen for BAV and ascending aortic disease in the first degree relatives of the patient with a BAV.
BAV may also be associated with an underlying syndrome associated with aortic and/or diffuse vascular disease. Familial thoracic aortic aneurysm and dissection (FTAA/D) related to ACTA2 mutations may be associated with BAV. This disorder may be associated with cerebral aneurysms, livedo reticularis, and patent ductus arteriosus. Loeys–Dietz aneurysm syndrome, due to mutations in TGFBR1 and TGFBR2, may also be associated with BAV. It is important to consider the presence of such underlying disorders when evaluating the individual with BAV and thoracic aortic aneurysm, especially when features suggesting these conditions are present.
When should prophylactic aortic surgery be performed in BAV disease?
The timing of prophylactic aortic root replacement associated with BAV disease is a complex decision. Many issues are at play including the absolute size of the aorta, the condition of the BAV, and the rate of expansion of the aortic aneurysm. The patient's age, gender, body size and weight, family history, underlying condition, and general health also play important roles. The risk of more complex aortic surgery must be weighed against the long term risk of aortic complications. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines for the management of patients with valvular heart disease include recommendations for the management of ascending aortic disease associated with BAV (box 2).17
ACC/AHA guidelines for managing bicuspid aortic valve with dilated ascending aorta
Class I
Patients with known bicuspid aortic valves should undergo an initial transthoracic echocardiogram to assess the diameters of the aortic root and ascending aorta. (level of evidence: B)
Cardiac MRI or cardiac CT is indicated in patients with bicuspid aortic valves when morphology of the aortic root or ascending aorta cannot be assessed accurately by echocardiography. (level of evidence: C)
Patients with bicuspid aortic valves and dilatation of the aortic root or ascending aorta (diameter >4.0 cm*) should undergo serial evaluation of aortic root/ascending aorta size and morphology by echocardiography, cardiac magnetic resonance, or CT on a yearly basis. (level of evidence: C)
Surgery to repair the aortic root or replace the ascending aorta is indicated in patients with bicuspid aortic valves if the diameter of the aortic root or ascending aorta is >5.0 cm* or if the rate of increase in diameter is 0.5 cm per year or more. (level of evidence: C)
In patients with bicuspid valves undergoing AVR because of severe AS or AR (see sections III-A-6 and III-B-2-g), repair of the aortic root or replacement of the ascending aorta is indicated if the diameter of the aortic root or ascending aorta is >4.5 cm.* (level of evidence: C)
Class IIa
It is reasonable to give β-adrenergic blocking agents to patients with bicuspid valves and dilated aortic roots (diameter >4.0 cm*) who are not candidates for surgical correction and who do not have moderate to severe AR. (level of evidence: C)
Cardiac MRI or cardiac CT is reasonable in patients with bicuspid aortic valves when aortic root dilatation is detected by echocardiography to further quantify severity of dilatation and involvement of the ascending aorta. (level of evidence: B)
*Consider lower threshold values for patients of small stature of either gender.
Reproduced with permission from: Bonow RO, Carabello B, Chatterjee K, et al. ACC/AHA 2006 Guidelines for the Management of Patients With Valvular Heart Disease: A Report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients with Valvular Heart Disease). J Am Coll Cardiol 2006;48:e1–148.
AR, aortic regurgitation; AS, aortic stenosis; AVR, aortic valve replacement.
It is recommended that patients with BAV and ascending aortic aneurysms exceeding 4.5 cm undergo simultaneous aortic replacement at the time of aortic valve replacement.17 In this document, it states that surgery to repair the aortic root or replace the ascending aorta is indicated in patients with BAVs if the diameter of the aortic root or ascending aorta is >5.0 cm or if the rate of increase in diameter is 0.5 cm/year or more, and to consider lower threshold values for patients of small stature of either gender.17 These recommendations are based on consensus opinion by a group of experts and not by randomised trials.
It is clear that with increasing size of an ascending aortic aneurysm, the risk of aortic dissection increases. However, recent data regarding absolute size of the aorta at the time of imaging in acute aortic dissection are provocative and raise important points with regards to the unpredictable nature of aortic dissection. In the IRAD there were 68 cases of dissection in those <40 years old, including 9% with BAV and 12% with prior aortic surgery.5 The average size of the ascending aorta at the time of dissection in this group was 5.4±1.8 cm. In the Yale experience of 70 ascending aortic aneurysms associated with BAV, six patients (8.6%) suffered aortic dissection or rupture in follow-up, with the average size of the ascending aorta at the time of dissection measuring 5.2 cm.18
Individuals with thoracic aortic aneurysms are at risk for aortic dissection with increased risk of dissection and rupture as aneurysm size increases, but many aortic dissections occur in patients without notably dilated aortic dimensions.4 Of 591 type A aortic dissections in the IRAD, the maximum aortic diameter averaged 5.3 cm with 349 patients (59%) having aortic diameters <5.5 cm and 229 patients (40%) having aortic diameters <5.0 cm. In a series of 177 non-Marfan patients with tricuspid aortic valves with acute type A aortic dissection, 62% of patients had a maximum aortic diameter <5.5 cm at the time of dissection, 42% had a maximum aortic diameter <5 cm, and more than 20% had a maximum aortic diameter of <4.5 cm at the time of dissection.4
Thus, the absolute size of the aorta is not the only factor important in triggering acute dissection. Age, gender, body size, and rate of aortic growth also play a role. Indexing the aortic size to body surface area may better predict risk of aortic dissection than using absolute aortic size alone.4 19 Underlying genetic triggers may lead to dissection at differing aortic sizes. Mechanical factors and aortic curvature may also affect risk of dissection. The mechanisms responsible for individual susceptibility for acute dissection at a certain aortic size are poorly understood.
Patients with BAV must be followed for late aneurysm formation
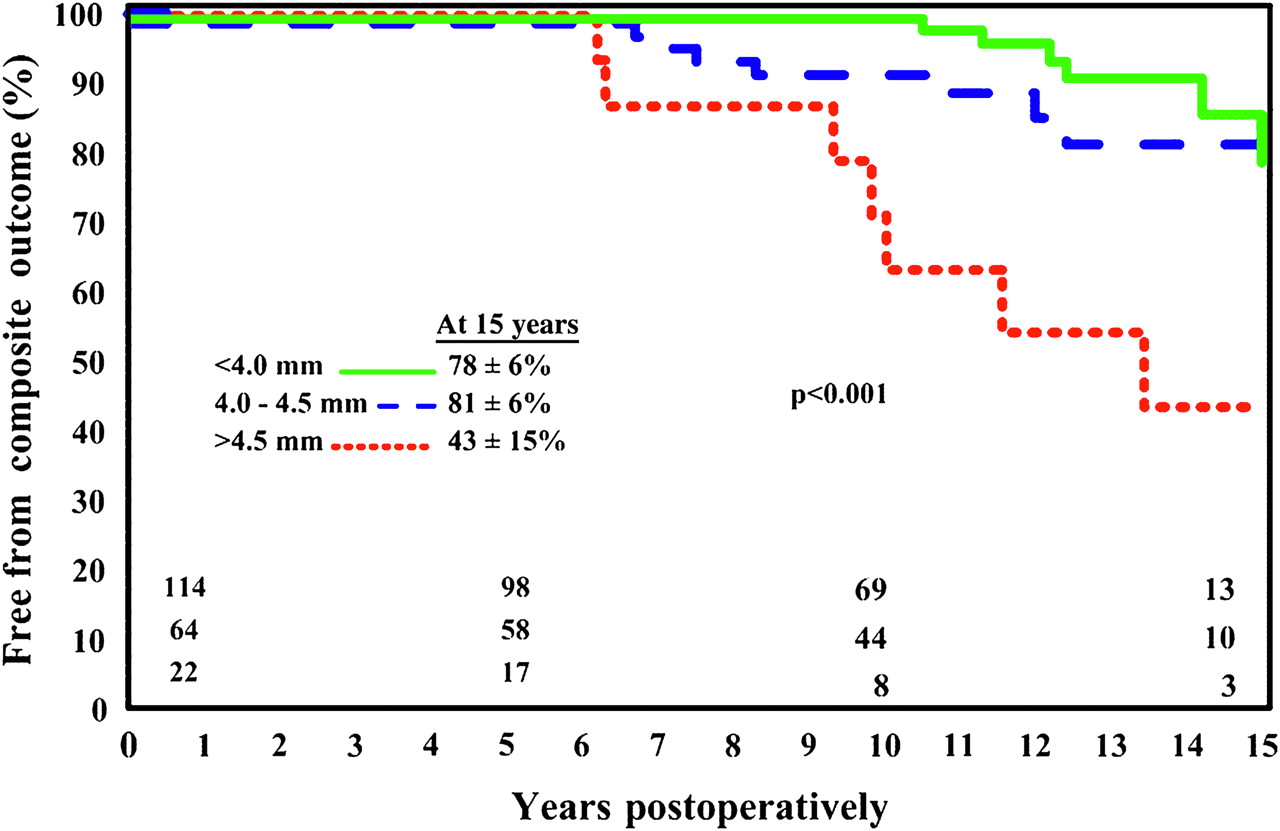
Late complications involving the ascending aorta, including aneurysm formation and aortic dissection, have been reported after aortic valve replacement (AVR) for BAV.1 Compared to patients who underwent TAV replacement, those who underwent AVR for BAV had a much greater risk of late aortic complications.1 In a 10 year follow-up study, Borger et al reported that 18 of 201 (9%) patients who underwent AVR for BAV required late ascending aortic replacement for aneurysm formation.20 In this study, all patients with aortic size >5 cm underwent aortic replacement at the time of AVR. Ascending aortic size was <4.0 cm in 57%, 4.0–4.4 cm in 32%, and 4.5–4.9 cm in 11% of patients. Freedom from ascending aortic complications, which included aortic dissection, aneurysm repair or sudden death, was 78%, 81%, and 43% for the three increasing aortic size groups, respectively (figure 8).20
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier curves for freedom from ascending aortic complications according to the three groups of patients with bicuspid aortic valves undergoing valve replacement surgery. Patients with an ascending aortic diameter of 4.5 cm or greater had a significantly increased risk of future aortic complications (aneurysm, dissection, or sudden death) (p<0.001). Reproduced with permission from Borger et al.20
Therefore, patients who undergo AVR for BAV require long term surveillance of the aortic root and ascending aorta. If the ascending aorta is not adequately visualised to determine its maximal dimension on echocardiogram, then CT or MRI is recommended.
Medical management in BAV aortic disease
There are many tenets to the medical management of patients with BAV disease and a dilated ascending aorta (box 3).
Management of bicuspid aortic valve (BAV) aortopathy
Identification by echocardiogram and CT or MRI scan.
At least annual surveillance of the aorta when aortic dilatation is present.
Screening of first degree relatives for BAV and ascending aortic disease.
β-blocker treatment for appropriate candidates.
Lifestyle modification involving physical activities.
Prophylactic aortic surgery when the aorta reaches 5 cm or at 4.5 cm when aortic valve replacement is indicated*.
Long term imaging of the aorta after valve replacement.
*Consider lower threshold values for patients of small stature of either gender, or when rapid aortic growth occurs or there is a family history of aortic dissection.
First, routine imaging studies with at least annual evaluation of the aortic valve and ascending aorta are important. When the aorta is 4.0–4.5 cm, I usually recommend annual imaging with echocardiography (and, if necessary, CT or MRI) to document stability. As stated earlier, if the entire extent of the ascending aortic enlargement is not adequately visualised by echocardiography, CT or MRI should be utilised. If the aorta is 4.5–5.0 cm, then I usually recommend imaging every 6–12 months, depending upon the individual patient and stability of measurements.
Because of the risk of aortic aneurysm formation, patients with BAV should receive information about this risk, and when significant aortic enlargement occurs, about the risk of aortic dissection. Guidelines for safe physical activities should be discussed and avoidance of strenuous physical activities and isometric activities, such as intense weightlifting, should be emphasised to the patient with BAV and aortic disease. Guidelines for participation in competitive athletics state that athletes with BAV without significant valve disease and aortic diameters <40 mm may participate in all sports, while those with aortic enlargement >45 mm are restricted to low intensity sports. Because high intensity weight lifting has been associated with aortic dissection, I caution all my patients with BAV against this activity, whether they have aortic dilatation or not.
Although β-blocker treatment is suggested (class IIa recommendation) for the patient with BAV and ascending aortic enlargement >4.0 cm (in the absence of severe aortic regurgitation), this is based on consensus opinion, and not based on any prospective data in this population.17 The rationale for this recommendation is based on the effect of β-blocker treatment in Marfan syndrome and the theoretical benefits of lessening aortic stress by β-blockade in BAV aneurysm disease.
In mouse models of Marfan syndrome, agents which block TGF-β signalling can attenuate aneurysm formation. Although there are very small studies demonstrating benefit of angiotensin receptor blocker and ACE inhibitor treatment in people with Marfan syndrome, at present there are no data from studies in animals or people demonstrating a similar benefit from these therapies in BAV aneurysm disease. Certainly, if hypertension is present, one may choose such an agent for treatment.
Conclusions
BAV is a common congenital defect which not only involves the aortic valve, but also affects the proximal aorta and may lead to complications including ascending aortic aneurysm and aortic dissection. The underlying pathophysiology of the aortic disease is most likely the result of an intrinsic defect involving the aortic media, which is probably genetically triggered. Aortic dilatation associated with BAV may occur independently of valvular dysfunction and may progress long after valve replacement. Proper management includes identification and long term surveillance of the aortic valve and aorta.
Because BAV and aortic disease may be familial, it is important to screen first degree relatives for this condition. I recommend that first degree relatives of the patient with a BAV and aortic aneurysm or a family history of aortic complications related to a BAV undergo an echocardiogram (and in some instances CT or MRI). The cost effectiveness of screening all first degree relatives of the individual with BAV has not been examined. However, auscultation for BAV is notoriously suboptimal, and echocardiography is necessary to evaluate effectively for BAV or associated aortic disease. For the patient with a BAV alone (and no aortic complications), I recommend the patient tell his/her first degree relatives about the potential for familial BAV. At a minimum, auscultation for valvular disease in these relatives should be performed.
Investigations continue into the basic genetic defects associated with BAV and its aortopathy and the cellular mechanisms and signalling pathways involved in this process. With understanding of these pathways, new treatments to attenuate this disease may be forthcoming.
Aortic involvement in patients with bicuspid aortic valve (BAV): key points
BAV is common, affecting ∼1% of the population.
BAV is associated with many different congenital heart defects.
BAV is associated with ascending aortic aneurysm and risk for aortic dissection.
BAV aortic dilatation is associated with cystic medial degeneration.
BAV aortic disease is associated with increased apoptosis and MMP activity.
BAV aortic aneurysm disease is progressive.
BAV aortic aneurysm formation may occur years after valve replacement.
You can get CPD/CME credits for Education in Heart
Education in Heart articles are accredited by both the UK Royal College of Physicians (London) and the European Board for Accreditation in Cardiology—you need to answer the accompanying multiple choice questions (MCQs). To access the questions, click on BMJ Learning: Take this module on BMJ Learning from the content box at the top right and bottom left of the online article. For more information please go to: http://heart.bmj.com/misc/education.dtl
RCP credits: Log your activity in your CPD diary online (http://www.rcplondon.ac.uk/members/CPDdiary/index.asp)—pass mark is 80%.
EBAC credits: Print out and retain the BMJ Learning certificate once you have completed the MCQs—pass mark is 60%. EBAC/ EACCME Credits can now be converted to AMA PRA Category 1 CME Credits and are recognised by all National Accreditation Authorities in Europe (http://www.ebac-cme.org/newsite/?hit=men02).
Please note: The MCQs are hosted on BMJ Learning—the best available learning website for medical professionals from the BMJ Group. If prompted, subscribers must sign into Heart with their journal's username and password. All users must also complete a one-time registration on BMJ Learning and subsequently log in (with a BMJ Learning username and password) on every visit.
References
- ↵
This book chapter is a state of the art review of BAV, encompassing the embryology, genetics, valvular and aortic complications, and outlines management strategies for this condition.
- ↵
This paper highlights the risk of aortic complications associated with coarctation of the aorta when a BAV is also present.
- ↵
This is a seminal paper emphasising the prevalence of aortic dilatation and risk of aortic dissection in patients with Turner syndrome. Of great importance is the recognition that the aorta in Turner syndrome patients is at risk of dissection at relatively small size and must be indexed to body size.
- ↵
- ↵
- ↵
This paper reported on the natural history of a large population of BAV patients reporting on the frequency of cardiac complications in BAV patients.
- ↵
This paper reported the course for BAV patients from a large congenital heart clinic in Toronto with over 20% of patients requiring intervention within 9 years of follow-up.
- ↵
- ↵
- ↵
This is one of the earliest papers demonstrating the association of aortic dilatation in patients with BAV, even when the aortic valve is functioning ‘normally’.
- ↵
This paper is an important work demonstrating the role of increased apoptosis in BAV aortas even in the absence of aneurysmal enlargement.
- ↵
This paper reported the differential expression of the proteolytic enzymes (MMPs) and their inhibitors in aortic aneurysm tissues of BAV and TAV patients. This imbalance may play an important role in aneurysm formation in BAV patients.
- ↵
- ↵
This paper highlighted the frequent genetic component of BAV, emphasising that BAV is often familial.
- ↵
This very important paper reported on several families in which BAV and ascending aortic aneurysm were inherited in an autosomal dominant condition. Of utmost importance was the finding that first degree relatives were frequently noted to have an ascending aortic aneurysm, whether or not a BAV was present.
- ↵
- ↵
- ↵
- ↵
- ↵
This large series reported the late development of aortic complications after BAV aortic valve replacement depending upon the preoperative aortic diameter and makes recommendations for when to replace the aorta in BAV patients.
Supplementary materials
Web Only Data hrt.2009.183871
Files in this Data Supplement:
Footnotes
Competing interests In compliance with EBAC/EACCME guidelines, all authors participating in Education in Heart have disclosed potential conflicts of interest that might cause a bias in the article. The author has no competing interests.
Provenance and peer review Commissioned; internally peer reviewed.