Article Text

Abstract
Objective Human cardiac ryanodine receptor 2 (RYR2) shows autosomal-dominant inheritance in catecholaminergic polymorphic ventricular tachycardia type 1 (CPVT1); however, de novo variants have been observed in sporadic cases. Here, we investigated CPVT1-related RYR2 variant inheritance and its clinical significance between familial and de novo cases.
Methods We enrolled 82 independent CPVT1 probands (median age: 10.0 (7.0–13.0) years; 45 male) carrying the RYR2 variants and whose biological origin could be confirmed by parental genetic analysis: assured familial inheritance (familial group: n=24) and de novo variants (de novo group: n=58). We examined the clinical characteristics of the probands and their family members carrying the RYR2 variants.
Results In the de novo group, the RYR2 variants were more likely located in the C-terminus domain and less likely in the N-terminus domain than those in the familial group. The cumulative incidence of the first cardiac events (syncope and cardiac arrest (CA) or CA only) of the probands at the age of 5 and 10 years was higher in the de novo group than in the familial group. Nearly half of the probands in both groups experienced CA events before diagnosis. Only 37.5% of their genotype-positive parents had symptoms; however, at least 66.7% of the genotype-positive siblings were symptomatic.
Conclusions CPVT1 probands harbouring de novo RYR2 variants showed an earlier onset of symptoms than those with assured familial inheritance. Cascade screening may enable early diagnosis, risk stratification and prophylactic therapeutic intervention to prevent sudden cardiac death of probands and potential genotype-positive family members.
- genetics
- epidemiology
- tachycardia
- ventricular
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
Variants of the human cardiac ryanodine receptor 2 gene (RYR2) are known arrhythmogenic underliers responsible for catecholaminergic polymorphic ventricular tachycardia type 1 (CPVT1),1–4 identified in ~60% of patients with catecholaminergic polymorphic ventricular tachycardia (CPVT)4–7 and clinically characterised as exercise-induced or emotional stress-induced polymorphic ventricular tachycardia (VT) capable of leading to sudden cardiac death, especially in young patients.8 The cardiac ryanodine receptor channel controls Ca2+ release from the sarcoplasmic reticulum to the cytosol during the plateau phase of the action potential responsible for myocardial contraction, and CPVT1-related RYR2 variants have been reported to cause abnormal Ca2+ leak from sarcoplasmic reticulum, which may induce arrhythmias under elevated adrenergic tone.9–11
Autosomal-dominant inheritance of the CPVT1-related RYR2 variants was initially identified from a large family cascade screening.1 4 De novo variants have also been found in 35%–92% of CPVT1 probands,12–14 but the phenotypic differences between de novo and familial cases remain unclear. Generally, mutation-specific genetic testing for family members is recommended if a disease-causative RYR2 variant is identified in the CPVT proband.15 16 However, limited evidence exists as to whether inheritance can be assessed by the phenotypes and who would benefit from genetic testing. Moreover, as genetic testing sometimes raises sensitive discussion and some family members are hesitant to undergo testing, further clinical data to support cascade screening are required. Hence, we aimed to investigate the inheritance of CPVT1-related RYR2 variants, their clinical significance between de novo and familial probands, and clinical features of family members in the familial group.
Methods
Settings and participants
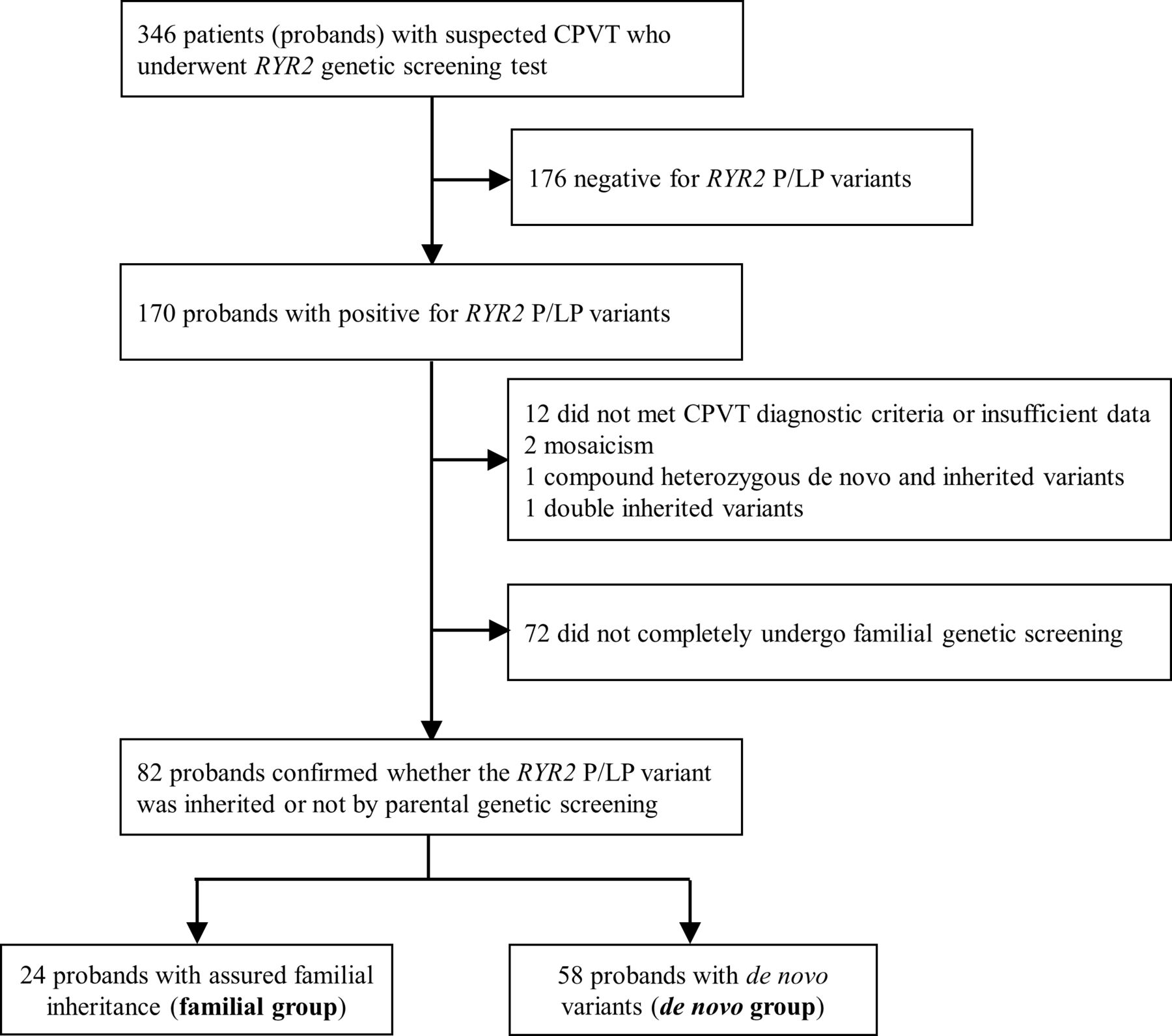
Among 346 Japanese patients (probands) with suspected CPVT and had undergone RYR2 genetic screening at the National Cerebral and Cardiovascular Center, Japan (2006–2021), or Shiga University of Medical Science, Japan, (2005–2020), 170 RYR2-negative probands and 13 probands who did not meet the CPVT criteria (n=12) or whose parents had an apparent mosaic RYR2 variant (n=2) were excluded. Probands who had a compound heterozygous variant inherited from both parents (n=1) and had a double mutation of maternal origin (n=1) were excluded. Furthermore, 72 probands whose family screening had not been completely performed were excluded. Therefore, in this retrospective study, we enrolled 82 sets of independent family lines whose probands were clinically diagnosed with CPVT carrying RYR2 variants based on the 2013 expert consensus recommendation (figure 1).8 For the RYR2 variants, we included only pathogenic or likely pathogenic variants according to the American College of Medical Genetics and Genomics guideline (online supplemental table 1).17
Supplemental material

Study profile. Study flowchart showing patients (probands) with CPVT with RYR2 pathogenic (P) or likely pathogenic (LP) variants and those in which RYR2 variants were determined to have or have not originated from either parents. CPVT, catecholaminergic polymorphic ventricular tachycardia.
Written informed consent was obtained from the subjects before genetic testing at each institute. Affirmative agreement for genetic and clinical studies from subjects and permission of their parents were obtained for child subjects. Some probands were previously described in studies from 2015, 2016 and 2018.14 18
When only one of the parents was identified as carrying the same RYR2 variant as the child (proband), we included him/her in the familial inheritance group, even if the other parent did not undergo genetic testing. Contrastingly, when neither parent carried the same RYR2 variant, we included them in the de novo group. In the inherited families, the genotype and phenotype of the siblings were also examined. The clinical characteristics of the de novo and familial groups, maternal-originated or paternal-originated variants, were assessed for all probands.
Clinical findings
Clinical information of the probands and family members were obtained from their medical records, and cardiac arrest (CA) and syncopal events were investigated. Syncope was defined as a transient loss of consciousness with or without seizures or confirmed ventricular arrhythmia that did not require resuscitation or defibrillation. The QT interval was corrected (QTc) for the heart rate using Bazett’s formula. We defined bradycardia as heart rate below the second percentile for age.19 20 Bidirectional VT was defined as beat-to-beat alternation of the QRS axis present for more than four beats on any ECG recording.21
Genetic testing and location classification
Genetic screening of all probands and families was performed by combining the conventional Sanger method, multiplex ligation-dependent probe amplification and next-generation sequencing using MiSeq (Ilumina, San Diego, California, USA), as previously described.18 Variants detected with next-generation sequencing were further reconfirmed by Sanger sequencing for accuracy in results. We assessed the RYR2 variant location distribution by classifying four groups based on the known disease-related variant cluster domain: (1) N-terminal domain (amino acid (AA) 44–466), (2) central domain (AA 2256–2434), (3) C-terminal domain (AA 3778–4201 and 4497–4959) and (4) others outside the three domains.9 22 23 As for missense variants, genetic significance was confirmed using information from public databases to exclude normal variation. This information was gathered in May 2021.
Statistical analysis
Quantitative variables are expressed as median (IQR). The Mann-Whitney U test was employed to compare continuous variables. Categorical variables are presented as number (n) and percentage (%) and compared using the χ² test or Fisher’s exact test. To evaluate eccentricity distribution of RYR2 variant locations, multiplicity-adjusted p values were calculated using the Bonferroni procedure. Gray’s test was used to examine the equality of cumulative incidence for first syncope, first CA and any first cardiac event before CPVT diagnosis in probands among the familial and de novo groups, and multiplicity-adjusted p values were calculated using the Bonferroni procedure. We treated diagnosis and medication therapy initiation as competing risks in order to analyse the probands’ event rate and diagnosis of probands or their siblings to analyse the siblings’ event rate. The OR with a 95% CI was estimated using univariable and multivariable logistic regression analyses and adjusted for sex to evaluate parent-predictive factors responsible for CA events in a proband. Results with a p<0.05 based on a two-sided test were considered statistically significant. Statistical analyses were performed using EZR (V.1.51; Saitama Medical Center, Jichi Medical University, Saitama, Japan),24 a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria) and SAS software (V.9.4; SAS Institute, Cary, North Carolina, USA).
Patients and public involvement
Patients and the public were not involved in the design, conduct, reporting or dissemination plans of our research.
Results
RYR2 variants and clinical phenotypes of probands
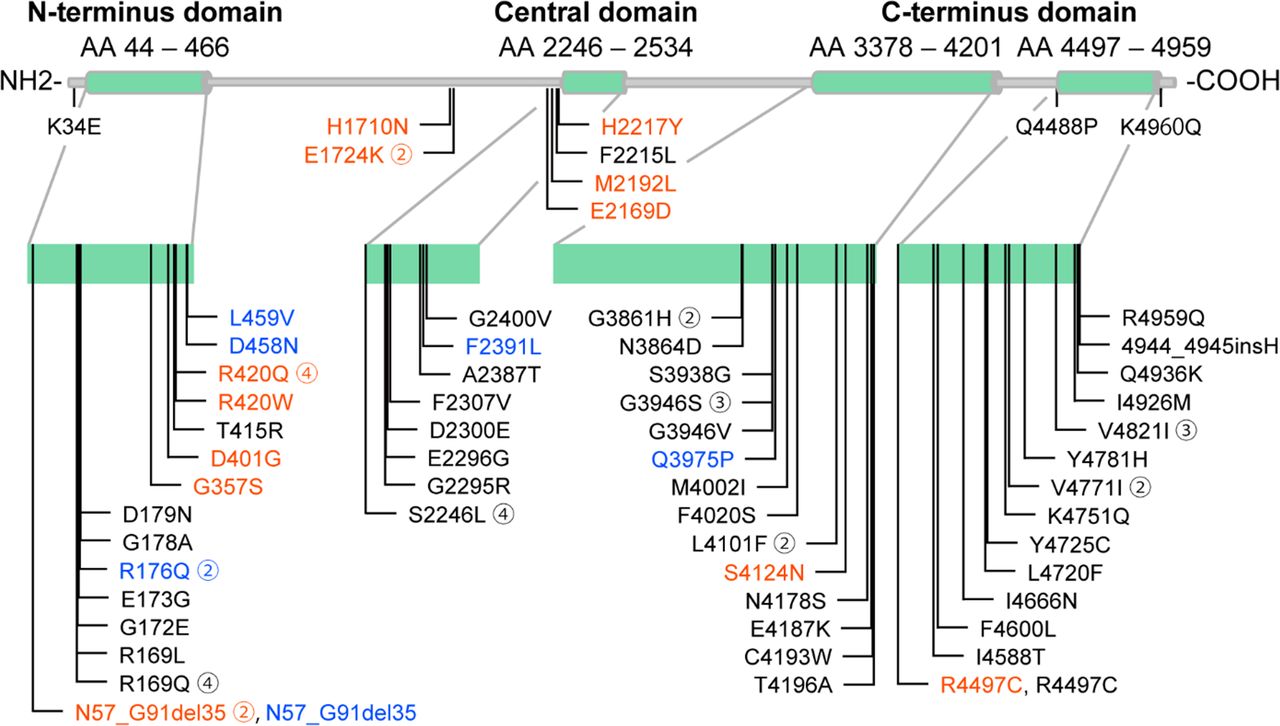
Among 82 CPVT1 probands, 45 (54.9%) were men, 61 (74.4%) had a history of syncope and 41 (50.0%) suffered from CA mainly triggered by exercise or emotional stress before diagnosis. Sixty-two RYR2 variants were identified, 59 of which were missense variants. The location of the RYR2 variants and the clinical background of each proband are shown in figure 2 and online supplemental table 2. Most of the variants were located in the N-terminus (AAs 44–466), central (AAs 2246–2534) and C-terminus (AAs 3378–4201 and 4497–4959) domains, which are CPVT1 hotspots.23 Information regarding missense variants, such as in silico prediction and allele frequency, obtained from public databases is shown in online supplemental tables 3 and 4.

Location of the RYR2 variants. The RYR2 variants of the probands in this study. Red characters, maternal-originated variants; blue characters, paternal-originated variants and black characters, de novo variants. The encircled number adjacent to each variant shows the number of probands who had the same variant.
Differences between de novo and familial probands
Fifty-eight probands formed the de novo group. Twenty-four probands were confirmed as the familial group, with the same RYR2 variants identified in the father (n=7) or mother (n=17). Details of the family pedigree of familial cases are shown in figure 3.

Family pedigrees of familial CPVT. Among 24 pedigrees of familial CPVT1 cases, 14 families underwent complete genetic screening for both parents. In the remaining 10 families, there was only one parent confirmed as genotype-positive, and the other was not completely confirmed as genotype-negative. CPVT, catecholaminergic polymorphic ventricular tachycardia.
The percentages of subjects with initial symptoms, either syncope or CA, and those with the worst symptoms before clinical diagnosis of CPVT did not differ between groups (table 1). However, age at occurrence of the first symptom, CA and clinical diagnosis were significantly lower, and bidirectional VT was more frequently documented in the de novo group than in the familial group. Neurological phenotypes such as epilepsy and intellectual disability did not significantly differ between the two groups and were not related to clinical phynotypes (table 1, online supplemental table 5). Distribution of variant location in RYR2 differed between groups (p<0.001), with variants in the de novo group more likely to be located in the C-terminus domain (33/57 (57.9%) vs 3/24 (12.5%), adjusted p<0.001) and less likely located in the N-terminus domain (10/57 (17.5%) vs 14/24 (58.3%), adjusted p<0.001) than those in the familial group.
Clinical characteristics and variant locations between de novo and familial probands
Figure 4 shows the cumulative cardiac incidence before diagnosis, and table 2 shows actual event rates at different ages and their differences between the groups. The cumulative incidence of syncope or CA was higher in the de novo group than in the familial group at 5 and 10 years of age. However, the total event rate at 15 years of age and the overall cumulative incidence were not significantly different between groups (adjusted p=0.36 and p=0.10) (figure 4A,B). The cumulative incidence of the first cardiac event was higher in the de novo group than in the familial group at 5, 10 and 15 years of age and cumulative incidence differed between groups (adjusted p=0.002) (figure 4C). This indicates earlier occurrence of the first event in the de novo group than in the familial group.

Cumulative cardiac incidence before diagnosis of CPVT in probands with RYR2 variants. Cumulative cardiac events of first syncope (A), first CA (B) and any of the first cardiac event (C) in probands harbouring RYR2 variants inherited from the parent or those with de novo cases. CA, cardiac arrest; CPVT, catecholaminergic polymorphic ventricular tachycardia.
Estimated cumulative cardiac incidence rate in probands and differences between de novo and familial cases at each age point
Effects of RYR2-variant-carrying parents on probands
Online supplemental table 6 shows the clinical characteristics of genotype-positive parents. Among 24 parents, including 7 (29.2%) fathers and 17 (70.8%) mothers, only 9 (37.5%) cases experienced syncope, including 1 (4.2%) concomitant history of CA. To investigate whether parental history of syncope or CA affected the total incidence of probands before diagnosis, we compared the probands with and without a parental history of syncope or CA (online supplemental table 7). Proband age at the first symptom was significantly lower in probands whose parents had a history of syncope or CA than in those without any symptoms in their parents (7.5 years vs 13.0 years, p=0.016). However, we observed no significant difference in other clinical features between groups. Logistic regression analysis revealed that parental clinical or genetic factors were not always associated with CA events in the CPVT1 probands (online supplemental table 8).
We further investigated how paternal or maternal origin affects the phenotype of probands, but we observed no significant difference in clinical findings between probands with paternal-originated or maternal-originated RYR2 variants (online supplemental table 9).
Clinical manifestation of genotype-positive siblings
All family pedigrees of the familial group are shown in figure 3. Two individuals had died at ages 17 and 19, respectively, (families 23 and 28, respectively) before genetic screening. Twenty-eight siblings belonging to the familial group (10 brothers and 18 sisters) underwent genetic testing, with 12 of them (3 brothers and 9 sisters) identified as carrying the same RYR2 variants as their probands.
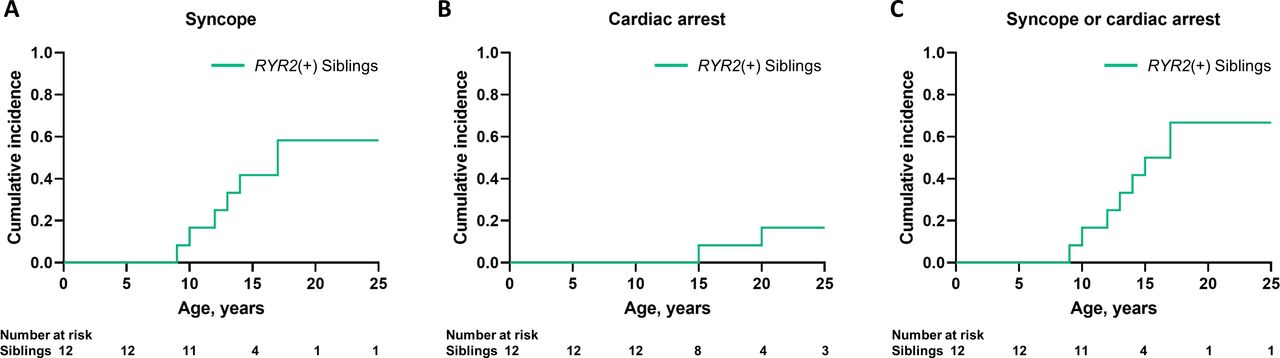
Among 12 siblings with the same RYR2 variants as the probands, 8 (66.7%) were symptomatic, 6 (families 3, 4, 22, 23, 24 and 52) had a history of syncope, 1 (family 4) had a history of CA and 1 (family 13) had experienced both CA and syncope. Figure 5 shows the cumulative first cardiac incidence of syncope (figure 5A) and CA (figure 5B) and any of the cardiac events (figure 5C) in genotype-positive siblings. None received medical treatment during genetic testing.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cumulative cardiac incidence in siblings carrying the same RYR2 variants as their probands. Cumulative cardiac events of first syncope (A), first CA (B) and any of the first cardiac event (C) in siblings harbouring RYR2 variants (RYR2(+)) inherited from their parents. CA, cardiac arrest.
Discussion
This is the first study that demonstrated complete trio genetic analysis of inheritance in CPVT1 probands. The main findings are the following: (1) age of any first cardiac event or CA was lower in the de novo group than in the familial group; (2) syncope comprised more than half of the first symptoms, and nearly half of the probands in both groups experienced a CA event before diagnosis; (3) one-third of genotype-positive parents were symptomatic; (4) two-thirds of genotype-positive siblings were symptomatic during genetic testing and (5) the locations of the RYR2 variants differed between familial and de novo group.
Relationship between inheritance and clinical manifestation of CPVT1
Among the CPVT-causative genes, RYR2 is the most common causative underlier and exhibits autosomal-dominant inheritance. Patients with CPVT1 are usually diagnosed by the age of 40 but can also be identified by genetic screening for idiopathic ventricular fibrillation.7 25 26 In the present study, >70% of the CPVT1 probands had a syncopal episode and ~50% had experienced CA before diagnosis. The age of event onset was lower in the de novo group than in the familial group, aligning with a previous study.14 Since many parents carrying the same RYR2 variants as probands were asymptomatic without any medication, the RYR2 variants in the familial group may be less pathogenic than those in the de novo group, resulting in a concealed phenotype or CA event at an older age. One presumption is that lethality prior to reproductive age may result in a more severe phenotype in the de novo cases than familial cases. Furthermore, more than half of the probands were de novo cases, which is relatively higher compared with the past report;2 however, we could not directly assess the familial/de novo ratio given the different genetic testing enquiry of parents between the studies. The popularity of large family cascade screening in CPVT-1 could have led to a more frequent diagnosis of the familial cases in the initial phase or a more aggressive recent genetic screening of asymptomatic families facilitated the diagnosis of the de novo cases.
RYR2 variants are associated with CPVT1 clusters in certain domains. Their structure-function analysis suggests that these loci are predominantly associated with intra-RYR2 domain interactions and cytoplasmic Ca2+-dependent channel modulation.2 An association exists between RYR2 variant location and the clinical phenotype of CPVT1, as patients harbouring variants in the C-terminus domain have an increased risk of non-sustained VT than those with variants in the N-terminus.12 However, the CA rate and age of the event in all probands did not differ between variants located in the N-terminus and C-terminus domains (online supplemental table 10). This suggested that variants within the same domain vary in severity and that the severity of each variant may determine its heritability.
Because mutational hotspots are largely related to loci prone to mutation during replication or DNA repair,27 it is reasonable to identify the same variant in different probands. However, in the present study, most variants did not overlap between groups, even if they existed within the same hotspots. This maldistribution may have resulted from different family lines having the same ancestral origin, whereas many of the variants found in the de novo group were not inheritable by the next generation.
Clinical implications of genetic screening for family members
Familial CPVT1 probands exhibited a late onset of symptoms than the de novo group; however, there were similar levels of CA risk if they had no proper medication before diagnosis, thus highlighting the difficulty in diagnosis without symptoms at any age. As a beta-blocker, flecainide and left cardiac sympathetic denervation can decrease the risk of life-threatening cardiac events in CPVT.3 13 26 28 An active attempt to diagnose and introduce early therapy would reduce CA risk. Although a burst-exercise test can reveal typical ventricular arrhythmias related to CPVT, an ECG cannot fully estimate cardiac event risk.29 30 Therefore, our results support the idea that cascade screening should be recommended for all family members, even if they have no symptoms.8 15 16 Indeed, genetic screening for asymptomatic families is sometimes a sensitive issue from the standpoint of potential ethical, emotional and social consequences of the test results, and careful genetic counselling and explaning the purpose of the examination are necessary before the genetic test.
Generally, relatives carrying the RYR2 variant exhibit a considerable phenotypic difference.12 In the present study, syncope or CA was only documented in 37.5% of genotype-positive parents, indicating that inheritance cannot be predicted based on symptoms. The symptomatic rate among genotype-positive siblings was relatively high (66.7%). The actual frequency may have been considerably higher if the two siblings who died before genetic diagnosis had been included. Additionally, because a previous study demonstrated that CPVT phenotype prevalence in family members increases up to 20 years of age,12 more asymptomatic genotype-positive siblings in the present study are expected to manifest the CPVT phenotype during the follow-up periods. Disease-modifier genes from the genotype-negative parent may also be a possible factor that results in phenotype presentation between the parents and their children. Thus, for children, early genetic screening may be strongly beneficial in preventing sudden cardiac death. Furthermore, considering the possibility of parents with genetic mosaicism, which was reported in one of the 63 patients with CPVT1 with RYR2 variants in a previous study,23 a genotype-negative result in both parents does not always guarantee the negative genotype in siblings. Therefore, comprehensive genetic screening should be recommended for all family members in order to enable early diagnosis and initiation of therapeutic intervention (online supplemental file 2).
Supplemental material
Limitations
First, only a small number of probands and their families were enrolled and there may be some selection bias in the enrolment. Several factors led to the small sample size of both groups, as de novo cases were difficult to be diagnosed, and familial cases were not included if their genotype-positive parents were deceased before cascade screening. However, given the lack of difference in rate of cardiac events rate, age of the events and bidirectional VT between the probands enrolled in this study and those excluded due to a lack of parental genetic test results (data not shown), selection bias based on the complete familial genetic screening would be limited. Second, cases of mosaicism cannot completely be ruled out by PCR-based Sanger sequencing30 and germline mosaicism by testing other organs. Third, not all probands who have had their RYR2 variants identified by Sanger sequencing underwent comprehensive genetic screening for other potential pathogenic variants. Fourth, not all probands and family members underwent exercise-stress testing to elucidate arrhythmogenic potential, especially the probands who had already suffered with VT or a ventricular fibrillation-storm episode, asymptomatic family members and younger children who could not undergo stress testing. Finally, this is a retrospective study and we could not fully follow up with patients after their diagnosis of CPVT1. As such, the prognostic difference between de novo and familial cases of CPVT1 after pharmacological and non-pharmacological therapies remains unclear. Further investigation with larger samples is required.
Conclusion
De novo CPVT1 cases demonstrated earlier onset of initial symptoms as compared with familial-inherited cases. Because two-thirds of the genotype-positive parents were asymptomatic and inheritance could not be predicted by their symptoms, genetic screening of parents and siblings in all CPVT1 cases may enable early diagnosis and prophylactic therapeutic intervention to prevent sudden cardiac death.
Key messages
What is already known on this subject?
Catecholaminergic polymorphic ventricular tachycardia (CPVT), an inherited arrhythmia that is potentially fatal in children, is difficult to diagnose, because the resting ECG is mostly normal.
RYR2 variants are identified in ~60% of clinically affected patients with CPVT, and genetic testing for probands and family members is recommended.
However, de novo variants are also identified in sporadic cases of CPVT probands and the phenotypic differences between de novo and familial cases of CPVT remain unclear.
What might this study add?
This is the first study demonstrating trio analysis of inheritance using a large number of CPVT probands.
CPVT probands harbouring de novo RYR2 variants as compared with those with assured familial inheritance showed an earlier onset of initial symptoms.
The distribution of the RYR2 variant location differed between the two groups.
Because not all genotype-positive parents were symptomatic and inheritance cannot be confirmed by parental symptoms, genetic screening of family members may help in risk stratification and early therapeutic strategies for CPVT.
How might this impact on clinical practice?
This study highlights the importance of why advancing genetic screening is necessary for families of CPVT probands.
To disclose the inheritance pattern, either de novo or familial, proband siblings should consider early genetic screening to prevent sudden cardiac death.
This study is clinically important for personalised risk stratification of patients with CPVT and families and particularly for child and adolescent health.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by M21-031-7, M29-167-6 (NCVC Ethics committee) and G2011-128 (Shiga Univ Ethics committee). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank Hiroaki Masuda, Kaori Kugo, Madoka Tanimoto, Arisa Ikeda and Kazu Toyooka for technical assistance with the genetic analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors All authors take full responsibility for data collection, analysis and interpretation; research conduct and manuscript submission. KS, SO, MH and TA contributed to study conception and design. SO, KS and NI performed genetic analysis. KK, KT, KS, MF, TM, YK, HS, TK, MW, KY, KI, YI, KM and SN collected the extracted data from patient records. SO and KA performed statistical analysis of all data. KS and TA drafted the manuscript, and SO, MH, TA and KK critically revised the manuscript. TA accepts full responsibility for the work as a guarantor.
Funding This study was supported by a grant from the Japan Agency for Medical Research and Development (AMED) (grant no. 17ek0109202 to SO and MH), a Health Science Research Grant from the Ministry of Health, Labour and Welfare of Japan for Clinical Research on Measures for Intractable Diseases (grant no. H27-032 to MH, SO and TA), Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (grant no. 15K09689 to SO) and Japanese Circulation Society research grant for genome analysis project in cardiovascular diseases (to TA and MH).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.