Article Text

Abstract
Objectives The prognostic value of genetic studies in cardiomyopathies is still controversial. Our objective was to evaluate the outcome of patients with cardiomyopathy with mutations in the converter domain of β myosin heavy chain (MYH7).
Methods Clinical characteristics and survival of 117 affected members with mutations in the converter domain of MYH7 were compared with 409 patients described in the literature with mutations in the same region.
Results Twenty-five mutations were evaluated (9 in our families including 3 novel (Ile730Asn, Asp717Gly and Arg719Pro)). Clinical diagnoses were hypertrophic (n=407), dilated (n=15), non-compaction (n=4) and restrictive (n=5) cardiomyopathies, unspecified cardiomyopathy (n=11), sudden death (n=50) and 35 healthy carriers. One hundred eighty-four had events (cardiovascular death or transplant). Median event-free survival was 50±2 years in our patients and 53±3 years in the literature (p=0.27). There were significant differences in the outcome between mutation: Ile736Thr had fewer events than other mutations in the region (p=0.01), while Arg719Gln (p<0.01) had reduced event-free survival.
Conclusions Mutations in the converter region are generally associated with adverse prognosis although there are differences between mutations. The identification of a mutation in this particular region provides important prognostic information that should be considered in the clinical management of affected patients.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Mutations in MYH7, encoding the β myosin heavy chain, are common causes of hypertrophic cardiomyopathy (HCM) and are also associated with dilated cardiomyopathy (DCM), LV non-compaction (LVNC) and restrictive cardiomyopathy (RCM). Initial reports proposed that the identification of specific variants in this gene would be useful in the prognostic evaluation of patients,1–6 a concept recently questioned. Our experience suggests it is useful for diagnosis, and provides prognostic information.
This hypothesis came from the evaluation of informative families carrying mutations in one of the most relevant functional domains of β myosin heavy chain: the converter region, located between amino acids 709–777 of the protein.7 Figure 1 summarises one of those families, described below, characterised by malignant disease with a marked phenotypical variability.

Phenotype of some carriers of the Gly716Arg mutation. A–C corresponds to the index case. (A) Left ventriculography showing marked trabeculations and invaginations. The echocardiogram (B) shows, in parasternal short axis view at mitral level in diastole, marked posterior trabeculations, also evident in the explanted heart (C). Images D–F correspond to the sister of the index case. The echocardiogram (D) shows asymmetrical septal hypertrophy in the systolic apical four-chamber view, with the typical ‘crescent moon’ shape of the LV and a mild systolic anterior movement of the mitral valve. In the parasternal short axis view at mitral valve level (E) we see septal hypertrophy and wall thinning of the inferoposterior LV wall. The explanted heart (F) shows that this thinned region corresponds with the presence of marked invaginations. Images G–I; correspond to the daughter of the index case. (G) parasternal short axis view immediately distal to the mitral valve shows posterior wall hypertrabeculation without hypertrophy at age 9 years. (H) Apical five-chamber systolic view when she was 13 years old with typical obstructive hypertrophic cardiomyopathy with a dynamic subaortic gradient of 64 mm Hg (I).
We hypothesised that mutations in the converter domain of MYH7 could be associated with severe disease expression and overlapping phenotypes, including HCM, DCM, LVNC and RCM. Additionally, different mutations in the same region would likely have different evolutions and prognoses, and their prognoses would be similar to that previously described for the same mutations.
Hence, our objectives were: (1) To evaluate and compare the clinical and prognostic implications of different mutations affecting the converter domain of the MYH7 gene, and (2) To compare the clinical course of our families with that described in the literature for carriers of mutations in the same region and for carriers of the same mutations.
Methods
We evaluated all the probands previously assessed in three specialised centres that carried mutations in the converter region. After obtaining an informed consent, genetic studies were performed by Sanger sequencing of coding exons and flanking intronic regions of the main sarcomeric genes, including MYH7, MYBPC3, TNNT2, TNNI3, TPM1, ACTC, MYL2, MYL3 and TNNC. Pedigree was drawn and genetic screening was offered to their relatives. A complete anamnesis, physical examination, ECG and echocardiogram were performed during the first evaluation of all relatives, prior to knowing the genetics result. The diagnoses of HCM, DCM, LVNC and RCM were done following the European Society of Cardiology and American Heart Association criteria.8–10 In deceased relatives the phenotype was assigned according to either clinical records or from information provided by the relatives. A diagnosis of ‘unspecified cardiomyopathy’ was assigned to cases with a previous diagnosis of cardiomyopathy, but incomplete information to define a specific phenotype (relatives either deceased or not accessible for our clinical evaluation). We considered as clinically affected and included in the survival analysis first degree family members without a genetic study who had died suddenly or due to heart failure under the age of 45 years. Studies were done after approval from the local Ethics Committees of the corresponding centres (Comité Ético de Investigación Clínica de Galicia-CEIC) and following the Helsinki declaration recommendations.
A comprehensive bibliographical search (PubMed, Web of Science and Google Scholar) collected all available clinical information of families and individuals who carried missense mutations within the converter domain (see online supplementary file references 1–100). We specifically reviewed documents with coincident authors to avoid the inclusion of duplicated cases. We included affected and unaffected individuals following the same criteria used for our cases. We assumed that deceased relatives who were considered clinically affected would be carriers of the mutation associated with the disease in their families.
Survival analysis
We defined the variable ‘cardiovascular death or transplant’ if one of the following events occurred: (1) unexplained sudden death, (2) heart failure death, (3) stroke death, (4) heart transplant, (5) appropriate implantable cardioverter defibrillator discharge or (6) death of unknown cause in patients younger than 45 years. For survival analysis we only considered families with available data in at least two relatives. Patients with known additional pathogenic mutations were not included in the survival analysis. We made the following comparisons:
Survival of our patients affected by different mutations in the converter region versus data in the bibliography on patients with the same particular mutations.
Cumulative comparison of the survival between two groups: previously published carriers of any mutation in the converter region versus carriers of any mutations in the converter region from our cohort.
Comparison of survival among different mutations in the converter region (combining our data with that previously published for the same mutations). We considered for this analysis those families with at least 10 individuals clinically evaluated.
SPSS (V.19.0) was used for descriptive statistics and survival analysis. The cumulative probability for the occurrence of the ‘cardiovascular death or transplant’ end point was estimated by using the Kaplan-Meier method and factors were compared by using the logrank (Mantel-Cox) method. Survival was calculated from birth. A two-sided p value<0.05 was considered statistically significant. Additionally, HRs with 95% CIs of the comparisons were estimated by a Cox proportional-hazards regression model including a frailty term that allows individuals to be clustered within families and the intercept to vary between families. This shared frailty model takes into account the possible correlation within the families.
This analysis was performed using R software, V.3.1.2. A two-sided p<0.05 was considered statistically significant.
Results
Description of the family with MYH7 Gly716Arg mutation
The index case was a 42-year-old man diagnosed with DCM. He also fulfilled criteria for the diagnosis of LVNC (figure 1A, B). Heart transplantation was performed due to refractory heart failure with severe systolic and diastolic biventricular dysfunction (figure 1C). His father had died of heart failure in his 40s, with a diagnosis of Chagas’ disease. His sister had been previously diagnosed with HCM (figure 1D, E) and she underwent heart transplantation at age 40 years for severe biventricular systolic and diastolic dysfunction. The explanted heart showed a thinned region with marked invaginations suggesting an overlapping phenotype between HCM and LVNC (figure 1F). The proband's daughter showed posterior wall and mild apical hypertrabeculation without hypertrophy at age 9 years (figure 1G). Four years later, she showed typical obstructive HCM (figure 1H, I). Mutation Gly716Arg was identified in all of them. The study by next generation sequencing of >200 genes previously related to inherited cardiovascular diseases in the two patients with transplant did not show additional mutations. Previous descriptions of families affected by this mutation had also shown overlapping phenotypes associated with a high incidence of premature sudden death, heart failure death or transplant (references in online supplementary file: 2–7,15,18, 19,21,23,27,34).
Evaluation of mutations in the converter region
We identified nine mutations in the converter region in 21 families (117 patients), with 81 proven and 36 probable carriers (table 1). Clinical diagnoses were HCM in 76, DCM in 2, LVNC in 1, unspecified cardiomyopathy in 7, SD in 16, and 15 were phenotype-negative carriers. Sixty-one (51%) experienced cardiovascular death or transplant. Table 1 summarises the distribution of families, patients, carriers and events for each mutation in our centres.
Patients and families with mutations in the converter region from our centres
Combining this information with that in the literature (table 2) results in a total of 25 different mutations identified in 526 patients from 157 families. Clinical diagnoses were HCM in 407, DCM in 15, LVNC in 4, RCM in 5, unspecified cardiomyopathy in 12, SD in 48 and 35 were healthy carriers. Ages of death and last follow-up were available for 334 cases. One hundred and eighty-four patients (37%) experienced cardiovascular death or transplant, including 111 sudden deaths, 27 transplants, 6 stroke related deaths, 32 heart failure deaths and 8 deaths of unknown cause at age <45 years. The median age at diagnosis was 18 years, range 1–75 years, 25th centile 9 years and 75th centile 34 years; 97% of the carriers were clinically affected by the age of 35 years.
Patients and families with mutations in the converter region from the bibliography
Three of the nine mutations from our cohort were novel (Ile730Asn, Asp717Gly and Arg719Pro). Thirty-seven of the 437 proven mutation carriers (8.4%) presented complex genotypes (double heterozygotes).
Survival analysis
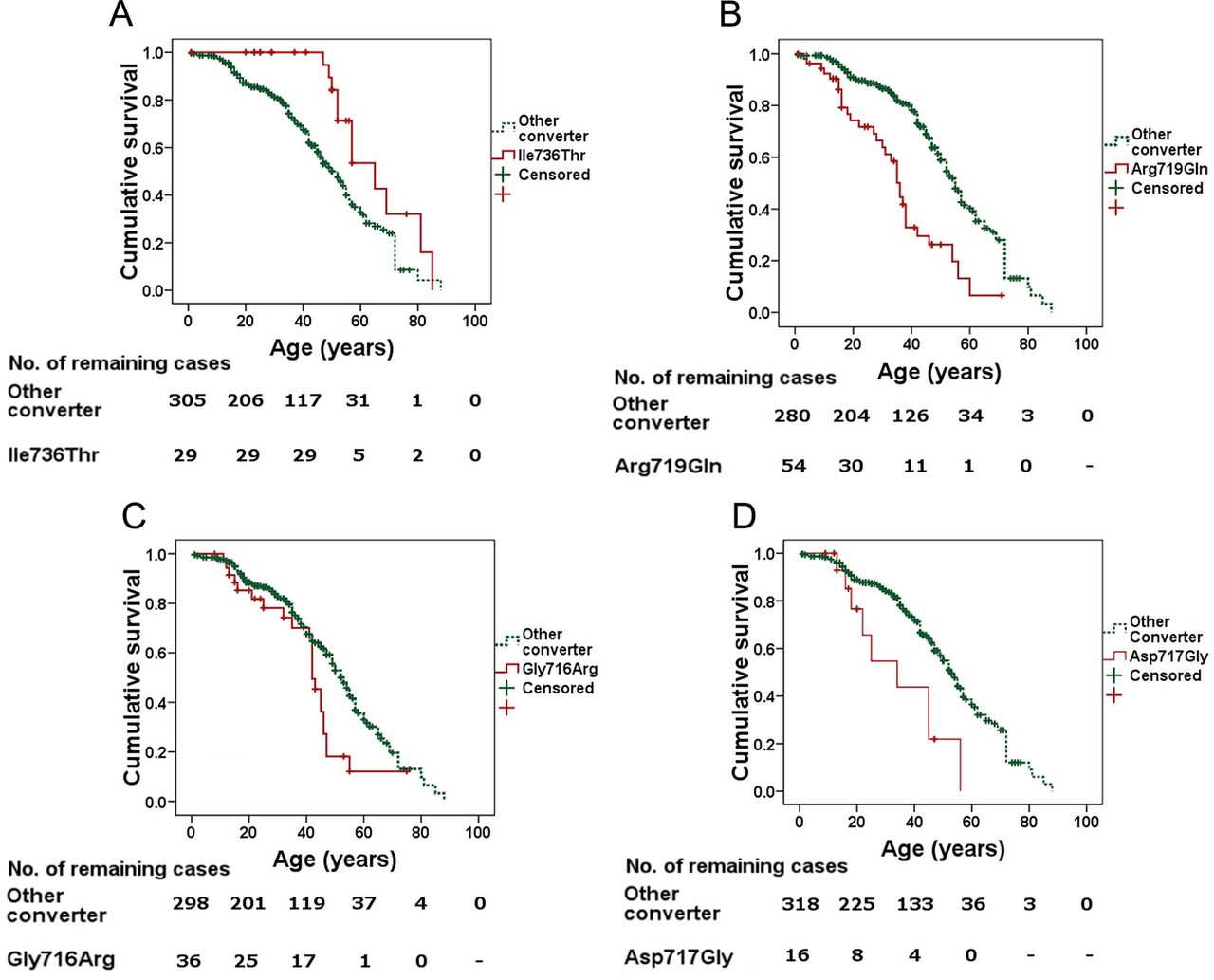
No significant difference in survival was observed between our cases with mutations in the converter region versus those from the bibliography (logrank test p=0.45; frailty model p=0.26, figure 2A and table A in online supplementary file). The median survival was 50±2 years in patients from our centres and 53±3 years in patients reported in the bibliography. There were no significant differences in survival between our patients with mutations Arg719Trp, Gly716Arg, Ile736Thr (figures Ia–c and table A in online supplementary file) and Gly768Arg (table A in online supplementary file) compared with those from the literature. Regarding mutation Arg719Gln, survival was similar for those under 50 years but the curves started to diverge after the age of 40 years as the two remaining patients from our centres showed a better survival after this age (logrank test p=0.03, frailty model p=0.02, figure Id and table A in online supplementary file).

Survival for all mutations: (A) Comparison between prognoses for all published mutations that matched with mutations from our centres. (B) Representative survival curves for four mutations in the converter region.
There were statistically significant differences in survival among different mutations (figures 2B and 3). Among mutations with at least 10 evaluated individuals, Ile736Thr showed a better prognosis than the rest of mutations (logrank test p=0.008, frailty model p=0.01) (figure 3A, and table A in online supplementary file). Kaplan-Meier survival curve showed a better survival for mutation Arg723Gly (logrank test p=0.031, figure II in online supplementary file), however statistical differences were not significant using the frailty model (p=0.08; table A in online supplementary file). Mutation Arg719Gln showed a worse prognosis than the rest (logrank test p<0.0001, frailty model p<0.01, figure 3B and table A in online supplementary file). Although Kaplan-Meier survival curves also showed a worse prognosis for the adjacently located mutations, Gly716Arg and Asp717Gly (logrank test p<0.01 each, figure 3B–D), differences were not significant for Gly716R and were in the limit of significance for Asp717Gly using the frailty model (p=0.28 and p=0.053, respectively). The same comparisons for mutations Arg719Trp, Arg723Cys, Gly741Arg, Gly741Trp and Gly768Arg showed differences not statistically significant using both statistical methods (p>0.1). Mutations with a worse prognosis were also characterised by an earlier age at diagnosis/onset, namely, Asp717Gly: median 10 years (range 2–14 years); Arg719Gln: 9 years (1–42 years) and Gly768Arg: 17 years (1–34 years). On the contrary, mutations having better survival rates had a later manifestation of the disease (Ile736Thr: 40 years (15–68 years); Arg723Gly 59 years (13–72 years)).

{kind=link}
{kind=link}
{kind=link}
Descriptive Kaplan-Meier curves for selected mutations. Comparison of carriers of a particular mutation versus carriers of all other mutations in the converter region. Mutation Ile736Thr (A) showed a better prognosis than the other mutations. Arg719Gln (B), Gly716Arg (C) and Asp717Gly (D) showed a worse prognosis than the rest of the mutations in the region.
No statistically significant difference in survival was observed in carriers of one versus two mutations (p=0.08; figure III and table A in online supplementary file).
Discussion:
The main findings of this work were:
Mutations in the converter region are, in general, associated with an adverse outcome, and the disease course in our patients with mutations in the converter region was comparable with the outcome of previously described patients with the same mutations.
Prognosis varies significantly between different mutations affecting the converter region.
A significant proportion of mutations (9 out of 25) was associated with more than one phenotype. This was observed among and within families, and multiple patients showed overlapping phenotypes (HCM, DCM, LVNC and RCM).
Prognostic value of mutations in the converter region
Initial reports tried to establish the prognostic value of several β myosin mutations.1 ,2 ,6 The term ‘malignant’ or ‘benign’ mutations emerged and after that, several groups of investigators continued in this line.4 ,11 However, subsequent studies questioned these results and the overall utility of genetic testing in prognosis. Mutations initially classified as ‘malignant’ or ‘benign’ showed different behaviours in other families and even in members of the same family.11 ,12 Moreover, mutations are usually found in isolated and/or small families, making it difficult to evaluate their prognostic implications. These considerations have discouraged the study of individual mutations and most studies have focused on the study of the prognostic differences between genes or functional domains.13
The results of our study support the concept that there is a relationship between prognosis and the affected functional domain; but we also show that within the same domain different mutations have different prognoses, which is probably related to the diverse structural implications and/or functional changes caused by each mutation. We demonstrate the high risk associated with mutation Arg719Gln (and probably Gly716Arg, and Arg719Trp mutations), which is also shared by the closely located novel Asp717Gly mutation. By the age of 50 years, only 20% of carriers of Gly716Arg, Arg719Gln or Asp717Gly are still alive, whereas about 90% of carriers of mutations Ile736Thr or Arg723Gly survive to this age. However, the slope of the survival curves for Ile736Thr or Arg723Gly mutations was similar to that shown by the most severe mutations, 20 years earlier; meaning that the 5-year annual risk of patients with the less severe mutations of the converter domain is quite high after age 40–50 years. Mutations Gly768Arg and Gly741Arg showed an intermediate prognosis, with just over 60% of patients alive at the age of 50 years (figure 2B). We could hypothesise from these results that amino acids 716–719 of the converter region could be critical for the protein function.
An original and relevant aspect of our study is the comparison by survival analysis with previous descriptions in the literature with the findings in our families for the same mutations. This has been possible because we have been able to collect a sufficient number of individuals for several of the identified mutations.
The disease course usually differs between carriers of a particular mutation, due to the influence of multiple genetic, epigenetic and environmental factors. Thus, the identification of the mutation provides information about the probable course of the disease, but does not absolutely determine the behaviour in each of the carriers. We suggest that the identification of a high-risk mutation in a patient should be considered together with classical clinical risk factors to establish the most appropriate therapeutic and preventive management. Some of the mutations we have described are associated with a high number of severe events in previously undiagnosed patients including children. In addition to triggering a timely family screening and follow-up of the carriers, the available information should be considered in the reproductive counselling and as a relevant factor for the decision on the indication of implantable cardioverter defibrillator for sudden death primary prevention in selected patients.
A second mutation is identified in 5–10% of cohorts of patients with HCM undergoing genetic studies, and classically indicates a more adverse prognosis. Our analysis failed to reproduce this finding. The presence of one particularly harmful mutation could reduce the potential role of the second variant. Alternatively, second mutations on top of a converter region mutation could have resulted in very severe phenotypes with fetal demise.
Limitations of the study
Clinical and genetic data on some of the relatives of the reported cases were not accessible. Some of the patients were diagnosed as ‘unspecified cardiomyopathy’ because the phenotype was not appropriately described in the available medical records or by the relatives.
In survival analysis we assumed that the cause of death in those patients with a premature event was related to the familial disease; something that might not be true in some cases. However, only two cases in the entire cohort presented this characteristic, and exclusion of these cases did not change our results.
For some comparisons of specific mutations between our cases and the literature and also when we compare specific mutation with the rest, the number of events and cases is low and the statistical power is insufficient to definitively reject the null hypothesis (absence of difference in survival). This limitation is accentuated when we use more exigent models that take into account the possible correlation within the families.
We are aware that referral and publication biases are likely to occur. However, referral bias would be minimal in our cohort as we receive and study patients diagnosed with the disease regardless of its severity, and in fact, most of the patients followed up in our clinic show a benign prognosis with an annual sudden death rate of approximately 0.5%.14
Conclusion
The identification of a particular mutation within the converter region of the β myosin heavy chain protein provides relevant information about the disease prognosis. Mutations in this region are associated with a particularly adverse outcome. They are usually related to the development of a severe form of HCM and also to overlapping phenotypes such as DCM, RCM and LVNC.
The prognosis does not significantly vary between different families with the same mutation but varies among different mutations within the region. Mutations such as Ile736Thr or Arg723Gly have a better outcome while Gly716Arg, Arg719Gln and Asp717Gly have an ominous prognosis. Identification of a high-risk mutation within this region should lead to proactive consideration of primary prevention of sudden death and close follow-up.
Key messages
What is already known on this subject?
Mutations in MYH7, encoding the β myosin heavy chain, are common causes of hypertrophic cardiomyopathy and are also associated with dilated cardiomyopathy (DCM), LV non-compaction (LVNC) and restrictive cardiomyopathy (RCM). Evaluation of mutation located in the same functional domain has been previously performed but sample size was too small to draw convincing conclusions. Prognostic value of mutations has been previously proposed but it has been recently questioned.
What might this study add?
Mutations located in a particular functional domain (converter domain of β myosin heavy chain) are associated with a particularly adverse outcome. They could determine overlapping phenotypes of hypertrophic, dilated, RCM cardiomyopathies and LVNC, even within the same family. The prognosis does not significantly vary between different families with the same mutation but varies among different mutations within the region.
How might this impact on clinical practice?
Identification of a high-risk mutation within this region should lead to proactive consideration of primary prevention of sudden death and close follow-up.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors DG-G: Acquisition of data, analysis or interpretation of data, planning, conduct and reporting. MA: Acquisition, analysis or interpretation of data, planning, conduct and reporting. MO-G: Acquisition of data. RB-V: Acquisition of data. XF: Acquisition of data. IR-G: Acquisition of data. AM: Acquisition of data, proofreading. EV: Acquisition of data. EM: Acquisition of data. PR: Acquisition of data. IL: Acquisition of data. LC: Acquisition of data. DF: Acquisition of data. JRG-B: Acquisition of data, proofreading. CS: Acquisition of data. JS: Acquisition of data. WM: Analysis or interpretation of data, planning. LM: Acquisition of data, analysis or interpretation of data, planning, conduct and reporting.
Funding This work has been supported by grants from the Carlos III Health Institute, FIS 2011: PI11/02604, FIS 2008: PI081834and RIC (Cardiovascular Research Network: Inherited Cardiovascular Diseases). The results of this work have been funded by the Project N° PI11/02604, integrated in the National Plan for Scientific Research, Development and Technological Innovation 2008–2011 and funded by the ISCIII- General Subdirection of Assesment and Promotion of the Research – European Regional Development Fund (FEDER) “A way of making Europe”; FIS 2008: PI081834, and RIC (Cardiovascular Research Network: Inherited Cardiovascular Diseases). The “Red de Investigación Cardiovascular (RIC)” of the Spanish Ministry of Health supports Barriales-Villa (RD 12/0042/0069), Fernandez (RD 12/0042/0069) and Gimeno-Blanes (RD 12/0042/0049).
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.