Article Text

Abstract
Pulmonary hypertension (PH) is a condition of multiple aetiologies with underestimated prevalence and incidence. Indeed, despite access to modern therapies, pulmonary hypertensive vascular disease (PHVD) remains a progressive, usually life-limiting condition, severely impacting on the patients’ well-being. We herein provide practical, expert consensus recommendations on the initial diagnostic work-up, clinical management and follow-up of children and adolescents with PH/PHVD, including a diagnostic algorithm. The major topics and methods that need to be tailored and put into context of the individual patient include PH classification, clinical signs and symptoms, basic diagnostic and advanced imaging measures (ECG, chest X-ray, transthoracic echocardiography, cardiac magnetic resonance, chest CT angiography, cardiac catheterisation, ventilation-perfusion lung scan, abdominal ultrasound), lung function tests, 6 min walk and cardiopulmonary exercise testing, sleep study (polysomnography), laboratory/immunological tests, considerations for elective surgery/ general anaesthesia, physical education and exercise, flying on commercial airplanes, vaccinations, care of central intravenous lines and palliative care. Due to the complexity of PH/PHVD, the clinical care has to be multidisciplinary and coordinated by a dedicated specialist paediatric PH centre, not only to decrease mortality but to allow children with PH/PHVD to reach a reasonable quality of life.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Video abstract
Box 1: Definitions
Pulmonary Hypertension (PH)
mPAP ≥25 mmHg in children >3 months of age at sea level
Pulmonary Arterial Hypertension (PAH)
mPAP ≥25 mmHg
PCWP <15 mmHg
PVR index >3 WU × m2
Idiopathic PAH (IPAH)
PAH with no underlying disease known to be associated with PAH
Hereditary PAH (HPAH)
PAH with no underlying disease but with positive family history or positive genetic testing
Pulmonary Hypertensive Vascular Disease (PHVD)
For biventricular circulations:
mPAP ≥25mmHg and PVR index >3 WU × m2
For circulations with cavopulmonary anastomosis (e.g., Fontan physiology):
mean TPG >6 mmHg (calculate mPAP minus mLAP or PCWP) or PVR index >3 WU × m2 Detailed hemodynamic definitions of PH (e.g., precapilary vs. postcapillary PH, value of the diastolic transpulmonary pressure gardient) are presented in Apitz C, Hansmann G, Schranz D. Heart, 2016[1]. mPAP, mean pulmonary artery pressure; mLAP, mean left atrial pressure; PAH, pulmonary artery hypertension; PCWP, pulmonary capillary wedge pressure (syn. PAWP, pulmonary artery wedge pressure); PVRi, pulmonary vascular resistance index; TPG, transpulmonary pressure gradient.
Introduction
Pulmonary hypertension (PH) is a condition of multiple aetiologies, sharing a significant increase in pulmonary artery pressure. For the majority of patients, PH represents a progressive and life-limiting disease with significant impact on quality of life, especially in the advanced stages of disease.
Diagnostic algorithms and implementation of careful disease monitoring and follow-up are desirable to standardise medical care, gain further insight into disease progression, guide therapy, facilitate multicentre trials and, ultimately, achieve best possible clinical outcomes. Consensus standardisation is particularly important because PH, especially pulmonary arterial hypertension (PAH), is a rare condition and single-centre experience is often limited.1 ,2
The aim of this article is to provide an overview over the condition and existing classifications. Moreover, we introduce a paediatric diagnostic algorithm and practical recommendations for the monitoring and outpatient care of children and adolescents with PH, PAH and/or pulmonary hypertensive vascular disease (PHVD).
Methods
The recommendations on diagnostics, monitoring and outpatient care in paediatric PH given in table 1 are based on a grading system currently suggested by the European Society of Cardiology (ESC) and the American Heart Association (AHA), and was based on paediatric data only (class of recommendation, level of evidence). The grading and voting process within the writing group is outlined in the executive summary69 of this special issue. Computerised searches of the PubMed/MEDLINE bibliographic database from 1990 to June 2015 were conducted. The developer searched using the terms ‘paediatric pulmonary hypertension’, ‘pulmonary hypertension and children’, ‘pulmonary hypertension and children and: diagnosis/diagnostics; follow-up, monitoring, epidemiology, classification, symptoms, functional classification, electrocardiogram, imaging, chest X-ray (CXR), echocardiography, lung function test, six-minute walk test, cardiopulmonary exercise testing, polysomnography, sleep study, computed tomography (CT), cardiac catheterization, magnetic resonance imaging (MRI), ventilation-perfusion scan, abdominal ultrasound, portal hypertension, chronic thromboembolic pulmonary hypertension, laboratory testing, serological markers, biomarkers, genetic testing, (lung) transplantation, palliative care, general anaesthesia, risk, surgery, flying, air travelling, contraception’.
Recommendations on diagnosis, monitoring and outpatient care in children with suspected or confirmed PH/PPHVD
Definition
PH has been defined as a mean pulmonary artery pressure (mPAP) ≥25 mm Hg at rest, measured by cardiac catheterisation (according to the most recent 5th World Symposium on Pulmonary Hypertension, Nice 2013). The term pulmonary arterial hypertension (ie, group 1 PH) describes a subpopulation of patients with PH, characterised haemodynamically by the presence of pre-capillary PH, including an end-expiratory pulmonary artery wedge pressure <15 mm Hg and a pulmonary vascular resistance >3 Wood units.3 ,4 While this definition has been widely accepted for paediatric use,5 the Pulmonary Vascular Research Institute (PVRI) expanded it and introduced the term ‘paediatric pulmonary hypertensive vascular disease (PPHVD)’ in 2011 (Panama, 2011). Importantly, the Panama classification distinguishes between PH with and without pulmonary vascular disease (PVD), between single and biventricular circulations, and acknowledges the heterogeneous aetiology in paediatric PH that can even have prenatal (fetal) origins. Although patients with congenital heart disease (CHD) and single-ventricle physiology (without subpulmonary ventricle) often do not meet the criteria as defined above (ie, mPAP <25 mm Hg, but PVD evident), they may benefit from similar pharmacological strategies and share some clinical features of patients with PH. For patients with Fontan-type haemodynamics (no subpulmonary ventricle), a PVR index >3 WU×m2 or a transpulmonary gradient of >6 mm Hg has been suggested as a definition of PPHVD.4 The most common type of PH diagnosed in childhood is PAH associated with CHD. Frequently, PAH-CHD presents with a pre-tricuspid or post-tricuspid shunt lesion (eg, left-to-right shunt) with or without evident PVD. Children without PVD benefit from shunt closure.70 Children with established PAH and PVD (PPHVD) may not tolerate shunt closure due to the sudden loss of decompression of the RV. The onset and progression of PAH-CHD and consecutive PVD differ in pre-tricuspid versus post-tricuspid lesions since the latter (eg, a large ventricular septal defect (VSD)), may expose the pulmonary vasculature not only to increased flow but also systemic or near-systemic pressure.
While adults with classical Eisenmenger haemodynamics actually have a better survival than patients with idiopathic pulmonary arterial hypertension (IPAH)/hereditary pulmonary arterial hypertension (HPAH), children with PAH-CHD and those with IPAH have a similar mortality with a 5-year survival reported to be 71% and 75%, respectively.6
PH classification
Nice World Symposium classification (2013) and Panama PVRI classification (2011)
Whereas the first international classification for PH was based on histological findings,7 more recent classifications (Evian 1998, Venice 2003 and Dana Point 2008) were refined with a focus on common clinical features, pathophysiology and to delineate differences and similarities of the various entities of the disease.
At the last World Symposium of Pulmonary Hypertension in Nice (2013, table 2), a new subgroup of ‘multifactorial PH’ was introduced to account for the heterogeneity of paediatric PH and special considerations that apply to children.8
Classification of pulmonary hypertension (5th World Symposium on Pulmonary Hypertension, Nice 2013)5
The Paediatric Taskforce of the PVRI addressed the need for a more specific paediatric classification, introducing the concept of hypertensive vascular disease (PPHVD), that is, PH with elevated PVR, and focusing on issues and disease entities that are particularly relevant to children.4 In 2011, a new Paediatric Panama classification was proposed (table 3), suggesting 10 categories of PPHVD. The Panama classification takes perinatal maladaptation, insufficient development and pulmonary hypoplasia, as well as genetic findings into account, all of which may play a role in the development of PH early in life. It also introduces a class of ‘multifactorial PH’ because—especially in children—often more than one factor may account for the diagnosis of PH. The main aim was to provide a system that is clinically useful, incorporates specific paediatric aspects that were previously not adequately addressed and allows categorising complex patients with a multifactorial pathogenesis of their PH accordingly (table 3).
Panama classification of Paediatric Pulmonary Hypertensive Vascular Disease (PPHVD) (2011): 10 basic categories of PPHVD
Epidemiology
Data on the exact incidence and the prevalence of PH in infancy and childhood are sparse. PH in childhood is seen with a variety of conditions, however, if at all, limited data are mainly available for IPAH and PH associated with CHD (PH-CHD). Two Dutch nationwide registries of paediatric PH suggested an annual incidence of 0.7 cases per million children for IPAH and a point prevalence of 4.4 per million children. For PH-CHD, annual incidence and prevalence were 2.2 and 15.6 cases per million children, respectively. Compared with adult studies, the incidence of IPAH seems to be lower in children, whereas PH-CHD may occur more frequently in childhood.9 Data from the UK Pulmonary Hypertension Service reported a slightly lower incidence for IPAH, that is, 0.48 cases per million children per year for IPAH and a prevalence of 2.1 cases per million children (IPAH).10 A 5-year multicentre prospective study including 21 referral centres in France reported an estimated prevalence of 3.7 paediatric PAH cases per million, and for IPAH, 2.2 cases per million.11
All in all, based on the available data a PH prevalence in 2–16 per million children can be estimated, acknowledging that there are risk groups in whom occurrence of PH in childhood and adolescence is substantially higher. This includes, for example, schistosomiasis,2 and probably other unrecorded, underserved children in developing countries with left heart disease leading to pulmonary venous hypertension, i.e. group 2 PH, for example, rheumatic heart disease (with mitral stenosis).
Diagnostic algorithm
Ideally, children with suspected or confirmed PH should be referred to, comprehensively evaluated and treated in multidisciplinary, specialist paediatric PH centres. The full assessment of invasive haemodynamics (right and left heart catheterisation), including ventricular performance and advanced non-invasive imaging, should be performed prior to the initiation of therapy by means of echocardiography,71 cardiac catheterization72 and cardiac MRI.73
However, the clinical status of the patient at the time of presentation has to be taken into account and stabilisation of the patient is paramount, even if this includes initiation of advanced PH-specific therapies before exact diagnosis.
In the process of the initial evaluation of ‘pulmonary hypertension’ and work-up, both a comprehensive medical history and physical examination are mandatory. Particular attention should be paid to accompanying symptoms and problems that could potentially suggest an underlying cause or modifier of PH (eg, prematurity— chronic lung disease; Raynaud's phenomenon—systemic sclerosis, systemic lupus erythematodes; Down syndrome—abnormal sleep study).
The diagnosis of IPAH can only be made after exclusion of other known factors playing a causative role in the pathobiology of PH applying a combined diagnostic approach for PH evaluation.
Basic diagnostics should include determination of the functional class (FC) of the child or adolescent (table 6). Further investigations, such as ECG, chest X-ray (CXR), echocardiography and functional testing (if feasible in that age), that is, lung function test, 6 min walk (6MWT) and cardiopulmonary exercise testing (CPET), should also be performed at the initial evaluation for PH.
Symptoms and clinical signs of pulmonary hypertension
Symptoms and clinical signs
Symptoms in PH are highly variable in individual patients and are often ‘non-specific’.5 ,12 During infancy and younger childhood, clinical presentation with overt right heart failure is extremely rare. Symptoms at rest (ie, WHO FC 4) are only present in very advanced disease. Young children may present with failure to thrive, school-age children often present with exercise-induced dyspnoea or dyspnoea at rest, syncope or chest pain.13 The diagnosis of PH may also be the result of coincidental findings (eg, increased cardiothoracic ratio on CXR, ECG abnormalities). Occasionally, children may be misdiagnosed with a bronchial illness or asthma. Possible clinical signs and symptoms at presentation in a child with PH are listed in table 4.
Physical examination should include the assessment of growth (percentiles on chart) in order to address any nutritional problems. Four limb blood pressure as well as four limb transcutaneous oxygen saturation (SaO2) should be obtained on initial assessment to detect any aortic arch/obstruction issues (eg, coarctation of the aorta), hypoxaemia or even differential cyanosis (eg, patent ductus arteriosus with predominant right-to-left shunt).
Functional classification
Functional classes (FC) are used to categorise patients into groups with comparable clinical status and disease-related compromise. FC often correlates with morbidity, quality of life and risk of death.14 The two most frequently employed functional classification schemes are the WHO and the New York Heart Association classification (table 5).15 ,16
WHO functional classification of pulmonary hypertension
Functional classifications are, however, not without problems, particularly in young children and children with handicaps (eg, Trisomy 21).
Therefore, a uniform, comprehensive and applicable classification with easily to determine variables would be desirable. The PVRI Paediatric Workforce recently suggested a specific functional classification system for different age groups, implementing factors such as the ability to thrive, developmental milestones and attendance of nursery/school (table 6).17 While promising, this newly suggested functional PH classification awaits external validation and correlation with paediatric outcome data. Despite the pitfalls using any functional classification, WHO FC evolved as one of few convincing prognostic factors in a recent meta-analysis.18
Paediatric functional classification of pulmonary hypertension
ECG
Right axis deviation beyond infancy (exceptions may exist in certain patients with PH-CHD), signs of right atrial dilatation, RV hypertrophy or strain, and significant intraventricular conduction delay can already provide important clues on the presence but not the severity of PH.
In patients with IPAH, RV hypertrophy was present in 87% and right axis deviation in 79% of patients on ECG.19 Due to the physiological right axis in younger children, this parameter is less reliable in infancy and in young children. A normal ECG for age, however, does not exclude the presence of PH.
Ventricular arrhythmias are rare in PH, but supraventricular tachycardias may occur. Atrial flutter or fibrillation may also be seen in advanced stages of paediatric PH.20 In PH patients with Eisenmenger syndrome, the occurrence of arrhythmia is thought to be associated with a poor prognosis.21
Taken together, for diagnosis, follow-up, decision-making or guidance of therapy, the ECG can be helpful; however, it cannot serve as a screening tool in isolation due to the limited sensitivity and specificity. However, given the easy access, low cost and wide availability, an ECG should be undertaken every 3–6 months.
CXR imaging
Ninety percent of adult patients with IPAH have an abnormal CXR at the time of diagnosis.19 Typical findings on CXR may be central pulmonary artery dilatation, pruning (suggestive of loss of peripheral blood vessels), as well as right atrial and RV enlargement in advanced cases.
A CXR usually allows to reasonably exclude moderate-to-severe lung disease or pulmonary venous hypertension due to left heart disease, leading to pulmonary oedema. However, the degree of PH does not necessarily correlate with the extent of radiographic abnormalities. In children with suspected PH, a CXR provides useful baseline information with acceptable risk/radiation exposure. Regular CXRs at follow-up are not indicated, unless there is a specific clinical indication (such as an underlying parenchymal lung disease).15
Echocardiography
While the definition of PH is still based on invasive haemodynamic measures, obtained by cardiac catheterisation, the first diagnostic tool and modality in suspected PH is generally the transthoracic echocardiogram.71
Echocardiography does not only allow a comprehensive initial assessment of cardiac anatomy, dimensions and function of both ventricles; it may also confirm the suspicion of elevated pulmonary pressures and usually allows for the quantification and estimation of those. Central venous pressure can be roughly estimated and functional parameters of RV and LV strain, contractility and ventriculo–ventricular interactions can be assessed.22
Despite the recognised limitations of echocardiography, to date it remains the imaging modality of choice that currently appears to be the most appropriate tool for longitudinal assessment of PH in children due to its non-invasiveness, wide availability and relative cost-effectiveness. Further details of a standardised approach including recommended parameters for the assessment of PH are discussed in greater detail in a dedicated article in this issue.71
Serial echocardiograms and ECGs are recommended every 3–6 months. In unstable or symptomatic patients or those who undergo therapeutic changes, more frequent echocardiograms may be indicated.
Lung function tests
In children old and mature enough to reliably cooperate, a basic lung function (or body plethysmography) test should be performed at the time of diagnosis. This is to rule out any underlying or coexisting airway or parenchymal disease (obstructive, restrictive or combined), which may need to be addressed independently from targeted PH therapy and warrant other treatment modalities. Static and dynamic parameters should be obtained, assessing presence and degree of restrictive or obstructive disease.
In patients with significant obstructive airway disease, symptomatic relief may be obtained from additional bronchodilator treatment. Furthermore, it is important to minimise respiratory comorbidity and avoid air trapping, consolidation or atelectasis, which often results in significant V/Q mismatch. The latter can negatively affect the underlying pathophysiology of PH aggravate PH-associated symptoms and hence contribute to confirm the severity of PH.
The presence of significant restrictive lung disease warrants a more detailed search for respiratory diseases involving the lung parenchyma and rule out interstitial lung disease, pulmonary veno-occlusive disease (PVOD) or pulmonary capillary haemangiomatosis (PCH), treatment of which may differ from advanced PH therapy; pulmonary vasodilator therapy can actually have adverse effects and may be contraindicated in PCH and PVOD.23 ,24
In addition, certain drugs (eg, inhaled iloprost) have been shown to cause bronchospasm in some children, and thus, a lung function test should be performed prior to the start of any inhalative PH therapy.25
6MWT and CPET
Serial CPET (treadmill, bicycle) and the 6MWT are recommended to assess and monitor exercise tolerance, gauge prognosis and for surveillance of therapy in children with PH of an appropriate age. Objective measures can be obtained and can be compared against normal values of healthy peers.
Unlike formal CPET, a 6MWT can be reliably performed from a young age (school children) onwards. Thus 6MWT represents an assessment tool for a wide age spectrum of patients. The 6MWT offers technical advantages versus CPET as it is easily applicable, does not require any expensive equipment and is less time consuming than a standardised CPET. Advantages of CPET include the greater variety of physiological measures, particularly additional information about respiratory capacity. In children with PH who are less compromised, routine CPET should be considered for an objective evaluation of their exercise capacity.26
During CPET, the following parameters should be obtained and have proven to be useful monitoring patients with PH: peakVO2, peak blood pressure and the ventilatory equivalent of CO2 (VE/VCO2 measured at ventilatory threshold or as slope) are of predictive value for survival and hospital admission. VE/VCO2- slope, which can be derived from submaximal exercise testing, has been shown to correlate with disease severity and mortality in a paediatric cohort with PH.27
In children with PH who still have a relatively preserved exercise capacity (eg, walking distance >300 m), there is no good correlation of 6MWT distance with peakVO2, while CPET provides a more accurate estimation of cardiopulmonary limitations to exercise capacity. However, in children whose 6MWTD is <300 m, peak VO2 correlates well with the distance walked. 6MWT may, therefore, actually serve as a test of maximal exercise in children with a greater degree of exercise intolerance.26 Box 2 outlines a standardised CPET ramp protocol for a sitting ergometer.28 For a comprehensive review of exercise testing in children with cardiac disease, see reference.29
Standardised cardiopulmonary exercise testing ramp protocol (sitting ergometer)
3 min rest on the ergometer
3 min warm-up with unloaded cycling (friction only), constant pedalling rate 60–70 min−1
Constant increase in work rate to exhaustion
Should be achieved in 8–12 min
Estimated from previous tests, body mass and anamnestic data:
Increase 5 W/min in very small children and severely disabled patients
Increase 10 W/min in patients with pulmonary hypertension (PH) and less affected children
Increase 15 W/min in very fit patients with PH
Increase 20–50 W/min in healthy adolescents and adults
3 min cool down with little load (about 1/10 of max. work load
At least another 2 min arrhythmia surveillance
From Cooper and Weiler-Ravell.28
Sleep study (polysomnography)
Patients with a suspicion or a predisposition for a breathing disorder (eg, patients with Down syndrome or with relevant daytime sleepiness, hypertrophic adenoids) and especially those with an inadequate response to PH-targeted pharmacotherapy should undergo a formal sleep study, which could provide additional information and may have direct therapeutic implications.30 ,31 Nocturnal CO2 retention does not only impact on the subjective well-being of the patient and functional capacity during daytime but may also aggravate PH/PHVD. If a sleep study is positive, appropriate measures such as counselling, the use of a sleeping mask or adenoidectomy in selected cases should be considered.32
High-resolution chest CT /CT angiography
Advanced chest CT imaging is recommended to exclude underlying parenchymal lung disease, PVOD, chronic thromboembolic pulmonary hypertension (CTEPH) and anatomical obstructions (eg, peripheral pulmonary stenosis or pulmonary venous stenosis), which may be beyond the diagnostic scope of routine transthoracic echocardiography.
A chest CT angiography is recommended in the initial assessment of children with suspected PH. However, if an obvious cause for PH/PHVD exists (eg, a large left-to-right cardiac shunt), these advanced imaging modalities associated with considerable costs and radiation may be omitted. A chest CT is the assessment modality of choice if parenchymal lung disease is suspected on CXR. In addition, a chest CT angiography is indicated in every patient being evaluated for lung transplantation.33
Cardiac catheterisation and cardiac MRI
A cardiac MRI (cMRI) study should be considered in children with suspected PH/PHVD as part of the initial diagnostic evaluation and during follow-up to assess changes in ventricular function, mass and volumes (chamber dimensions) in case it can be performed without sedation/anaesthesia. If sedation/anaesthesia is needed, the risk of sedation/anaesthesia needs to be balanced against the potential gain of information of the cMRI study and its impact on the future therapy of the individual patient with PH. In our experience, the risk of sedation/general anaesthesia is higher in patients with severe disease (history of pulmonary vascular crisis, syncope, high PVR, WHO FC 3–4) and those without a RV decompressing shunt e.g., IPAH.
Due to the important therapeutic implications, the complexity and specific risks and benefits of the procedures, separate articles in this issue are dedicated to cMRI73 and cardiac catheterisation72 including acute vasodilator testing.34–39
Ventilation/perfusion lung scan (V/Q scan)
A V/Q lung scan is more sensitive than CT in detecting peripheral thromboembolic disease, and thus, in adults, it is the method of choice to rule out potentially treatable CTEPH.40 Because CTEPH has a very low incidence in children compared with adults,41 a V/Q scan should only be considered in children when there is suspicion for the presence of CTEPH rather than using this method as a screening tool. However, if a definite diagnosis is still uncertain after exhaustion of other imaging modalities (cardiac catheterisation, high-resolution chest CT (HRCT)), a V/Q scan can be useful for selected patients.42
Abdominal ultrasound
Porto-pulmonary hypertension in childhood is rare;43 however, if no underlying cause of the PH/PHVD is evident, an abdominal ultrasound is indicated to rule out liver cirrhosis and/or portal hypertension. The use of contrast agents and colour Doppler have been reported to improve diagnostic accuracy in adults.43–46
Laboratory and immunological screening tests
It is unclear which laboratory tests are particularly useful in the diagnostic work-up of PH/PHVD in children as data are sparse: table 7 provides an overview of routine blood tests, which should be considered for the diagnostic work-up of adolescent and adult patients with PH at first presentation. For children, it appears reasonable to be more selective and to take the ethnic background, travel history and to accompanying symptoms into account. For example, although schistosomiasis is a frequent cause of PH worldwide, it is probably not necessary to test for it in most European children (table 7).42 ,47
Overview of routine laboratory blood tests in paediatric pulmonary hypertension
Genetic testing
Genetic counselling and testing is recommended for families with children with IPAH or HPAH. HPAH patients’ family members who develop new cardiorespiratory symptoms should be evaluated immediately for PAH.
For further details on comprehensive biomarker and genetic testing, please see the dedicated article in this issue.74
Diagnostic algorithm
The Tracking Outcomes and Practice in Paediatric Pulmonary hypertension (TOPP) registry investigators reported that to date, in the majority of children with PH, the current diagnostic work-up recommended for adults are not followed (figure 1). This finding highlights that adult guidelines do not appropriately meet the demands of a thorough work-up tailored for children and/or probably a lack of awareness of existing guidelines for adults.2 ,38

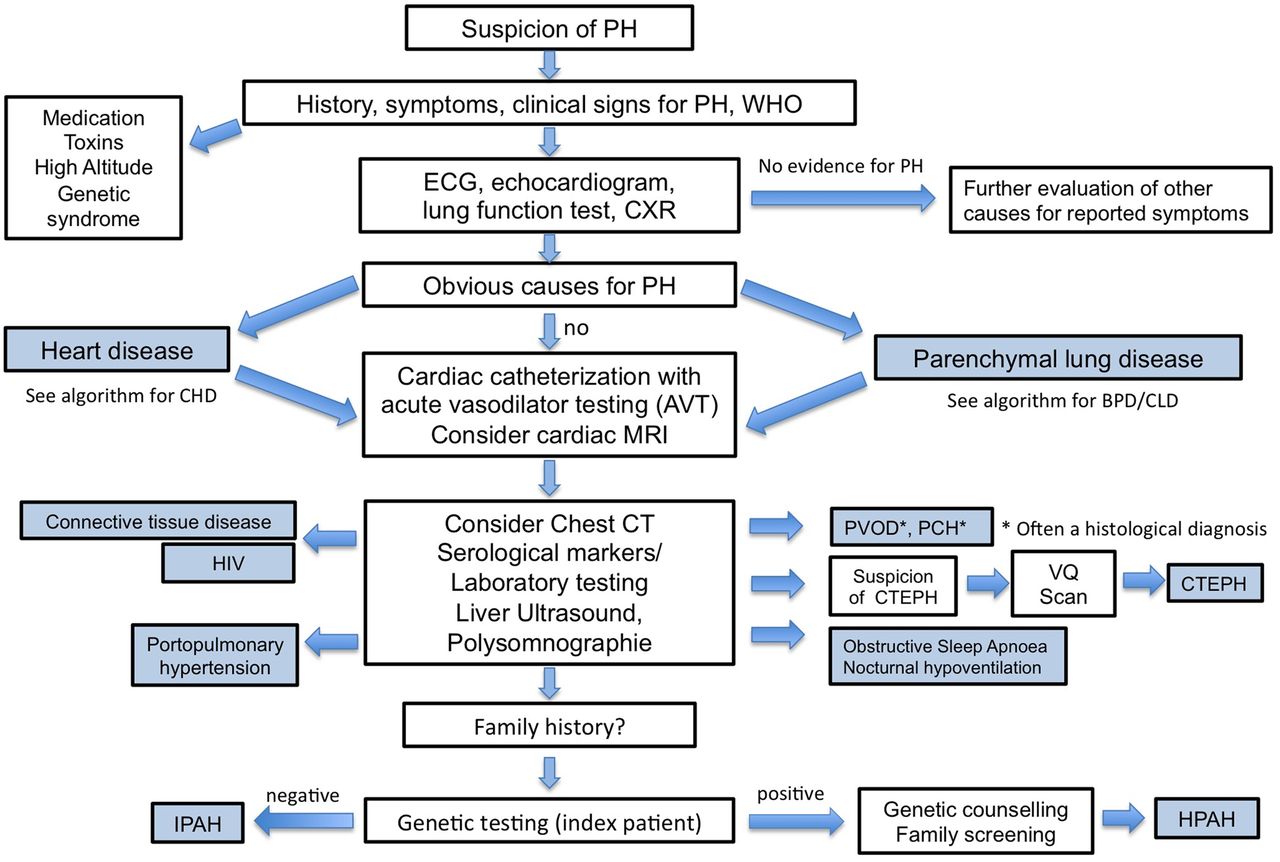{kind=link}
Diagnostic algorithm for a child or adolescent with suspected pulmonary hypertension. CHD, congenital heart disease; CPET, cardiopulmonary exercise testing; CTEPH, chronic thromboembolic pulmonary hypertension; CXR, chest X-ray; HPAH, hereditary pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; PCH, pulmonary capillary haemangiomatosis; PH, pulmonary hypertension; PVOD, pulmonary veno-occlusive disease; BPD, bronchopulmonary dysplasia; CLD, chronic lung disease; LFT, liver function test.
If PH/PHVD is severe and the patient presents severely ill in overt heart failure and/or pulmonary vascular crisis, cardiac catheterisation may be postponed due to its inherent risks and pharmacotherapy including intravenous prostanoids started immediately.72 ,75 Assessment may be postponed until the patient is clinically stabilised. A separate article in this issue discusses cardiac catheterisation and vasodilator testing in greater detail.72
Screening for paediatric PH is performed by ECG and echocardiography at the discretion of and after discussion among the primary paediatrician and paediatric cardiologist. If these investigations suggest the presence of PH/PHVD, CXR and HRCT should be considered.
Results from the TOPP registry revealed that ECG, echocardiography and CXR were the most frequently non-invasive tests used in the diagnostic work-up of children with PH. Of 456 patients included, none of them had all three tests considered normal, indicating a possible strength of combining these non-invasive tests to rule out PH with a reasonable degree of certainty.38 Once the combination of ECG and echocardiography points to the diagnosis ‘PH’, cardiac catheterisation, biomarker testing (NT-proBNP or BNP) and other tests should be performed, as outlined in table 7.
The TOPP registry revealed that once the diagnosis ‘PH’ was established, in <25% of children BNP and NT-proBNP testing was performed. 6MWT and CPET were conducted in only 38% and 7%, respectively.38
Finally, (combined right and left) cardiac catheterisation should be performed in all forms of significant PH (i) to confirm diagnosis and to distinguish between (ii) PH without elevated PVR and PH with elevated PVR (=PPHVD), (iii) pre-capillary from post-capillary PH/PPHVD, and (iv) to determine right atrial pressure, as well as RV and LV end-diastolic pressures as surrogate for diastolic function. Patients with PAH at diagnosis (and selectively at follow-up) need to undergo pulmonary vasoreactivity testing (for according recommendations, see article on cardiac catheterisation.72
If basic investigations including cardiac catheterisation,72 confirm the diagnosis of PH/PHVD, treatment should be initiated and an appropriate follow-up plan should be made for the individual patient.76
Monitoring and outpatient care
Table 8 shows an overview of investigations and tests that are recommended at the time of diagnosis and during follow-up, as well as additional optional tests that should be considered on an individual basis. Depending on clinical status, outpatient follow-up visits in 3-month to 6-month intervals appear reasonable. More frequent visits may be indicated in patients with progressive, advanced or end-stage disease, or after initiation or significant changes in therapy.
Recommended tests/investigations at diagnosis and follow-up of a child or adolescent with pulmonary hypertension/pulmonary hypertensive vascular disease
Patients on intravenous or subcutaneous prostanoid therapy should be seen at least every three months,48 ,49 whereas stable patients on oral (combination) therapy can be seen at 6-month intervals.
Some advanced therapies (eg, endothelin receptor antagonists) warrant further visits for laboratory testing.76
During every outpatient visit, a thorough history should be sought, and changes or occurrence of new clinical symptoms should be evaluated. Patients’ functional status and potential change of FC should be documented. Depending on children's age, maturity and cooperation, physical functional testing such as 6MWT and/or CPET should be performed in 6-month to 12-month intervals.
Elective surgery/general anaesthesia
Elective procedures under general anaesthesia require careful consideration of the underlying indication and judgement of the risk–benefit ratio as any general anaesthesia bares a substantial high risk for a child with moderate to severe PH. If a potential benefit of the procedure (gain of information) outweighs the estimated risk, or the surgery/intervention is unavoidable, timely planning is required. Management by an experienced anaesthesiologist who is familiar with PH haemodynamics and the involved pitfalls should be facilitated.
Good communication between teams, awareness of the severity of the disease and risks, and timely preparation are recommended. Conscious anaesthetic strategies maintaining an adequate systemic vascular resistance and adequate preload, and appropriate postprocedure monitoring are crucial to reduce morbidity and fatal procedure-related events.50–52
Pregnancy/Contraception
Pregnancy has a very high risk of fatal outcome for a pregnant woman with PH (30–50%) and for the fetus, and in the majority of cases, the caring physician will recommend termination of pregnancy. Female adolescents with PH/PPHVD should undergo timely counselling regarding options for secure contraception.53 ,54
Physical education and exercise
CPET testing is recommended for children with mild to moderate PH/PPHVD prior to engaging in any recreational or athletic activities.
It is recommended that children with stable mild to moderate PH/PPHVD should engage in light to moderate aerobic activity but be advised to self-limit their activities as required, avoid strenuous exertion and dehydration.
Paediatric patients with severe PH (FC III or IV and/or systemic or supra-systemic PA pressure) and/or recent history of syncope carry a high risk of sudden cardiac death with exercise and thus should not participate in any (competitive) sports.
Flying on commercial airplanes
Data on children exposed to cabin pressures and evaluation of need for supplemental oxygen are lacking. Generally, it is recommended that patients with PH should only fly on commercial airplanes in a stable and compensated condition. For patients with advanced disease, systemic pulmonary artery pressures and/or impaired ventricular function, it appears reasonable to use supplemental oxygen during the flight to minimise hypoxic vasoconstriction. The ESC and European Respiratory Society guidelines for PH recommend in-flight supplemental oxygen for patients with PH in FC III or IV and those with arterial partial oxygen pressure consistently <8 kPa (60 mm Hg).42 ,55
The PH patients’ caregivers should contact the airline in advance regarding the availability of supplemental oxygen in sufficient amounts during airline transport.
Vaccinations/Care of central intravenous lines
Children with PH should be under shared care and should be primarily seen and linked to a local paediatrician as first line of contact for all regular childhood investigations, routine vaccinations and general medical problems. However, good proactive communication and close cooperation is mandatory between specialised physicians seeing the patient with a PH focus and local community teams. Paediatricians should be actively encouraged to contact PH physicians for advice, for example, seeking support for and planning minor procedures, not necessarily related to the PH itself, where the patient could be exposed to any excessive risk (eg, general anaesthetic for dental procedure).
Children with PH should undergo all recommended routine vaccinations to prevent avoidable infections. Respiratory syncytial virus (<2 years of age), pneumococci and influenza vaccinations should be administered in paediatric PH patients if no contraindications exist.
Antibiotic prophylaxis for the prevention of subacute bacterial endocarditis is nowadays reserved for special patient subgroups (eg, cyanotic patients).56
In patients with a permanent central venous access (eg, for the administration of continuous prostacycline analogues), the caregivers must be vigilant for possible line infections. If clinically suspected (eg, unexplained fever, erythema or other infectious signs around line entry site), the continuous infusion of the prostanoid must not be abruptly disconnected. Due to the short half-life, the prostanoid has to be transitioned to a peripheral venous access, while the infusion via the central line is carefully weaned. Transition should ideally take place in a stepwise manner with monitoring of the clinical status, saturation and blood pressure of the patient, before blood cultures can be withdrawn from the central line. However, if line infection is confirmed, the chance to permanently clear the existing central line is often unsuccessful, despite antibiotic treatment. Most patients require removal of the infected line and alternative central line placement. Ideally, replacement should take place after a few days of antibiotic treatment and control of systemic infection/bacteraemia to avoid instant contamination of the new central line. The same technical process applies to patients with a leaking line or line dysfunction. They need to be transferred to the next emergency department immediately for intermittent peripheral intravenous line placement to ensure continuation of prostanoid therapy (epoprostenol has a very short drug half-life).
School
Close contact to healthcare providers and teachers at school should be sought. The aim is to familiarise them with practicalities of supporting a chronically ill child, to allow for a (near-) normal school attendance, to maximise pupils’ integration and to address understandable concerns and anxieties of the educators. Arrangements at school have to be made to facilitate the application/intake of any medication required during school hours (eg, inhaled iloprost, oxygen).
Additional interventional and surgical therapies
Atrial septostomy is often considered as the last opinion before active listing for bilateral lung transplantation. Indeed, atrial septostomy should represent a conscious step of active PH management particularly in patients with syncope.57 Paediatric experience is available and describes clinical improvement and abolishment of syncope, albeit an associated procedural risk, has to be acknowledged particularly for patients with increased right atrial pressures.58
Lately other surgical measures (Potts shunt) and interventional procedures (ductal stenting) have been introduced and offered to small numbers of patients.76 Paediatric experience is sparse and measures may be discussed as compassionate care. A detailed discussion of the involved risks and benefits is beyond the scope of this article; however, further details can be found in references.59–62 ,76
Lung transplantation
For some children, bilateral lung transplantation may be an option. However, despite improved immunosuppression regimens, overall survival after lung transplantation remains limited compared with other solid organ transplantation.
Median survival following paediatric lung transplantation is reported to be 4.9 years (2013 International Society of Heart and Lung Transplantation guideline).63 ,64 Acute rejection and bronchiolitis obliterans syndrome remain a major clinical problem responsible for both morbidity and mortality.65 Open counselling is important to balance risks and benefits and gauge whether this is a desirable and realistic option for each individual patient and their families. In patients with end-stage PHVD, early referral and evaluation by an interdisciplinary paediatric transplant centre should be sought.65–67
Palliative care
As disease progresses, it is important to support patients and families beyond providing high-quality medical care. Regardless of the decision to be actively listed for bilateral lung transplantation in end-stage disease, the extent of intensive care support should be discussed in a timely manner and alternatives, for example, palliative care, should be offered. Additional psychological support for families and patients and contact to professional social networks may be helpful.
General support
Children, siblings and caregivers of paediatric patients with PH/PHVD should be assessed for psychosocial stress and be readily provided psychosocial support and referral to appropriate institutions as needed. Individualised recommendations should be made, dependent on the underlying cause of the child’s PH and clinical status. Participation in physical education classes, access to education facilities and supporting aids (eg, wheelchair) to maintain mobility for patients with advanced disease should be discussed on an individual basis to meet the patients’ and families’ needs.
Conclusion
Due to the relatively low prevalence of PH/PHVD in childhood, the complexity of the disease, the heterogeneity of presentation and disease progression, and the need of a multidisciplinary approach, patient care and follow-up should take place at dedicated PH centres. In the setting of progressing disease and over time increasingly compromised patient with PH/PHVD, a balance has to be sought between regular visits at specialist PH clinics and good competent support from local medical teams, and palliative care services close to the patient's home.
References
Footnotes
This manuscript is a product of the writing group of the European Paediatric Pulmonary Vascular Disease (PVD) Network (Writing Group Chair: G. Hansmann, Writing Group Co-Chair: C. Apitz). ISHLT, International Society of Heart and Lung Transplantation; DGPK, German Society of Paediatric Cardiology
Funding AL receives grant support from the dean's office Medizinische Fakultät, Westfälischen Wilhelms-Universität Münster (15-002). CA currently receives grant funding from Stiftung Kinderherz (2511-10-13-001) and Behring-Röntgen-Stiftung (59-0018). GH currently receives grant support from the German Research Foundation (DFG; HA 4348/2-1), Fördergemeinschaft deutsche Kinderherzzentren (W-H-001-2014) and Stiftung Kinderherz (2511-6-13-011).
This Heart supplement was produced with support from an unrestricted educational grant from Actelion Pharmaceuticals Germany GmbH, Bayer Pharma AG, and Pfizer Inc. None of these organisations had any influence on the composition of the writing group or the content of the articles published in this supplement. Open Access publication of this article was sponsored by Actelion Pharmaceuticals Germany GmbH.
Competing interests AH received reimbursements for advisory board meetings from Actelion, and speaker's reimbursements from Actelion, Pfizer, Encysive, AOP Orphan Pharmaceuticals, OMT, GlaxoSmithKline; he received travel grants from Pfizer, GlaxoSmithKline, AOP Orphan Pharmaceuticals, Lilly, Actelion. His institution contributed to clinical trials sponsored by Actelion and Lilly. AH's institution received unrestricted scientific grants from Pfizer, Actelion and GlaxoSmithKline. AL, CA, PZ, KOD and GH declare no conflict of interest.
Provenance and peer review Commissioned; externally peer reviewed.