Article Text

Abstract
Objectives Paediatric pulmonary arterial hypertension (PAH) after neonatal arterial switch operation (ASO) for transposition of the great arteries (TGA) is a clinically recognised entity with an estimated incidence of 0.6%–1.0%. Nevertheless, a clinical characterisation is lacking. We present an international cohort of children with PAH after neonatal ASO for TGA and describe epidemiology and clinical course.
Methods Data were collected of children with PAH after neonatal ASO (≤6 weeks after birth) for simple TGA without residual shunt defects, identified in four national paediatric PAH networks in Europe and one US referral centre.
Results Twenty-five children were identified between 1989 and 2014. In 17 children (68%), PAH was detected <1 year after ASO. In the remaining children, PAH was detected after median 64 months (IQR 19.5, 94.5). Nineteen children (96%) received PAH-targeted therapies. During follow-up after ASO (median 5.2 years), eight children died, four underwent lung transplantation and two received a Potts shunt. 1-year and 5-year Potts shunt- and transplantation-free survival after ASO was 100% and 73%. From first PAH detection, this was 100% and 58%, respectively, which did not differ between children with early (<1 year after ASO) or late PAH detection.
Conclusions The occurrence of PAH after ASO for TGA represents a specific association. PAH onset may be early or late after ASO, with similar fatal course from first PAH detection. Mechanisms leading to PAH in this association are unknown, but may include abnormal prenatal pulmonary haemodynamics and/or genetic susceptibility. Routine, lifelong follow-up for children who undergo ASO for TGA should include screening for PAH.
Statistics from Altmetric.com
Introduction
Pulmonary arterial hypertension (PAH) is a rare and progressive disease of the pulmonary vasculature and has a poor prognosis.1 In children, PAH has an estimated annual incidence rate of approximately three cases per million children and is most frequently idiopathic or associated with congenital heart disease (CHD).1 ,2 CHD in PAH typically includes the presence or history of a shunt defect, as increased pulmonary blood flow is believed to trigger the remodelling of small pulmonary arteries that is characteristic for PAH.3 However, PAH has also been reported in children with CHD other than shunt defects.2 ,4
Transposition of the great arteries (TGA) is one of the most common cyanotic CHD, contributing to approximately 5% of all CHD.5 Early development of severe pulmonary vascular disease (PVD) has been well recognised in patients with uncorrected TGA.6 ,7 With its occurrence reported already in the first weeks of life, PVD in TGA seems to develop more rapidly than in other types of CHD.7 ,8
In earlier days, surgical management for TGA consisted of functional repair with an atrial redirection procedure (the Mustard or Senning procedures). These procedures were usually performed in the second half of the first year of life or even later, resulting in long-term presence of cyanosis and shunt lesions. Consequently, PAH is a well-recognised late complication of these procedures and has been reported to occur in approximately 7% of patients who survive into adulthood.9
Since the 1980s, the arterial switch operation (ASO) has become the treatment of choice for simple TGA.10 This procedure, in which the pulmonary artery and aorta are literally switched and the coronary arteries reimplanted, provides an anatomical repair and has a very good prognosis.11 The operation is usually performed within the first 2 weeks of life, precluding the presence of a long-term shunt lesion as a trigger for the development of PAH in patients with TGA.
Nevertheless, the association of PAH and TGA, also after a successful neonatal ASO, has been clinically recognised in paediatric pulmonary hypertension (PH) centres. An explanation for this association is unknown, but proposed mechanisms include programming of endothelial dysfunction due to prenatal hypoxic or postnatal hyperoxic blood perfusing the pulmonary vasculature in uncorrected TGA, genetic susceptibility, abnormal bronchial circulation and the dispersion of microthrombi, for instance, during atrial balloon septostomy.12–14
To date, however, a clinical characterisation of presentation, risk factors and prognosis of this entity is lacking. With the current study, we aim to characterise this clinical entity by presenting an international cohort of children with PAH after neonatal ASO for TGA and describing its epidemiology and clinical course.
Methods
This study is a retrospective, international multicentre study. Children with PAH and TGA repaired in the neonatal period with ASO between 1989 and 2014 were identified from nine dedicated paediatric PH centres in Europe and the USA, including four national registries (UK, France, Spain and The Netherlands). Ethical approval for the registries was obtained from the institutional review boards (of the constituent/participating registries), and the participants or their guardians provided written informed consent at enrolment.
Neonatal correction was defined as ASO within 6 weeks after birth. To exclude PAH associated with shunt defects, only patients with no haemodynamically relevant residual shunt defects after ASO were included in this study.
Patients and data collection
Patient characteristics were collected from patient charts, including gestational age, sex, medical history, the presence of other possible PAH-associated conditions and anatomical cardiac diagnosis. Only children with no haemodynamically relevant residual lesions were included, that is, only patients with isolated TGA or TGA associated with a ventricular septal defect (VSD) that had been successfully closed during ASO. Also, children with residual pulmonary branch stenosis or impaired left ventricular function were not included in this study. Parameters regarding the ASO and postoperative phase included whether there were any PAH-relevant complications and/or pulmonary hypertensive crises peri-ASO.
PAH was confirmed by cardiac catheterisation in all but one patient. PAH was defined as a mean pulmonary artery pressure ≥25 mm Hg with a mean pulmonary capillary wedge pressure ≤15 mm Hg. In one child, mean pulmonary capillary wedge pressure was not available at cardiac catheterisation, and another child had an echocardiographic PAH diagnosis. In both children, left heart disease was excluded by echocardiography on review of the centre's expert physician. The age of first PAH detection, either with echocardiography or cardiac catheterisation, was determined.
Treatment information included the use of supportive therapies, PAH-targeted therapies (including endothelin receptor antagonists, type 5 phosphodiesterase inhibitors and prostanoids) and surgical interventions during follow-up, that is, atrial balloon septostomy, Potts shunt or lung transplantation. Treatment intensity was defined as the number of PAH-targeted drugs at endpoint or last follow-up (PAH-targeted monotherapy, dual therapy or triple therapy).
Statistics
Data are presented as number (percentage) or median (IQR) when appropriate. The first occurrence of Potts shunt, lung transplantation or death was defined as the primary endpoint in this study. Patients who did not die nor received a Potts shunt nor underwent lung transplantation were censored at the last follow-up visit. Potts shunt- and transplant-free survival was depicted using Kaplan-Meier curves. Differences in survival were explored using the log-rank test. Values of p<0.05 were considered significant.
Results
Patients
In total, 25 children with PAH and neonatal ASO for TGA were identified (table 1).
Participating networks/centres and number of patients
All children were born after a gestational age of at least 36 weeks. Most children (76%) were males. Nineteen children (76%) had an intact ventricular septum, and six had a concomitant VSD (table 2).
Patient characteristics and ASO
Four children (17%) had a history of perinatal asphyxia. Three (13%) had associated persistent pulmonary hypertension of the newborn (PPHN), one of whom required perioperative extracorporeal membrane oxygenation. No recognised comorbidities, syndromes or dysmorphic features were reported, except for epilepsy and hydrocephalus associated with perinatal asphyxia in one child. No other causes for or conditions associated with PAH were identified in any of the children.
Twenty-one children (84%) underwent an atrial balloon septostomy in the first days of life. Median age of ASO was 8 days. Four children had a small (residual) VSD after ASO, considered not haemodynamically relevant.
At first cardiac catheterisation after ASO, median mean pulmonary artery pressure was 48 mm Hg and median indexed pulmonary vascular resistance was 11.5 Wood units m² (table 3).
PAH characteristics
Time of PAH detection
Median age at first PAH detection was 3 months (IQR 1, 14) with a range of 1–137 months (table 3). In fact, PAH was detected within 1 year after ASO in 17 children (68%) with a median age at first PAH detection of 1.5 months (IQR 1, 3). In the remaining eight children, median age at first PAH detection was 64 months (IQR 19.5, 94.5). Patient and disease characteristics, as shown in tables 2 and 3, did not differ between these two groups. All three children with PPHN had first PAH detection within 1 year after ASO.
Follow-up
Median follow-up after ASO was 5.1 years (IQR 2.9, 12.1). During follow-up, two children received a Potts shunt, four children underwent lung transplantation and eight children died. Of the eight deceased children, five died of progressive right ventricular failure, one of massive haemoptysis, and two died during a follow-up cardiac catheterisation, one of whom during atrial balloon septostomy procedure for PAH. Potts shunt- and transplant-free survival rates of 1, 3, 5 and 10 years after ASO were 100%, 82%, 73% and 65%, respectively (figure 1). From first PAH detection, these were 100%, 73%, 58% and 50%, respectively.

Potts shunt- and transplant-free survival from ASO of children with PAH after neonatal ASO for TGA. Survival of 1, 3, 5 and 10 years after ASO was 100%, 82%, 73% and 65%, respectively. ASO, arterial switch operation; PAH, pulmonary arterial hypertension; TGA, transposition of the great arteries.
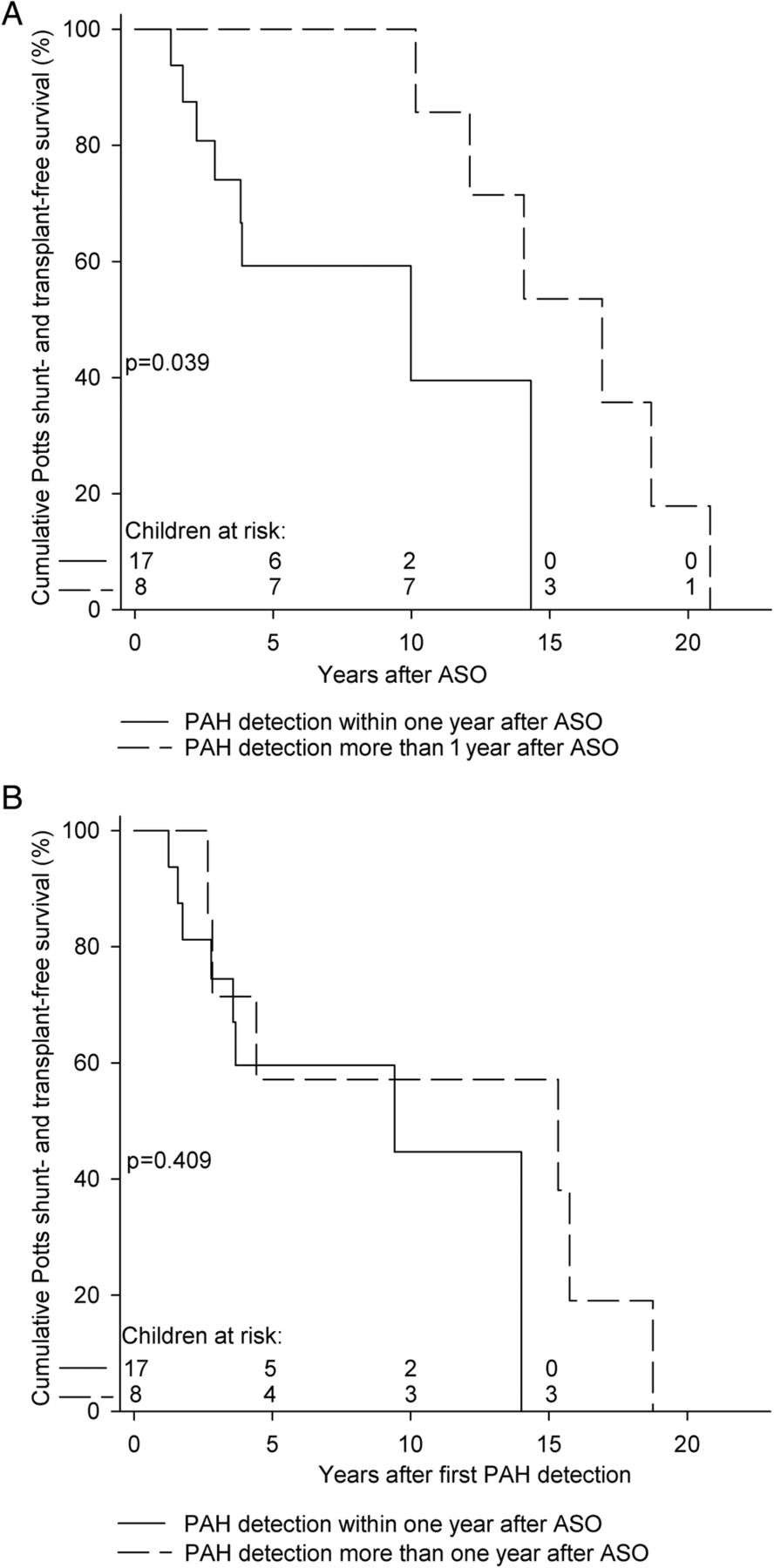
Potts shunt- and transplant-free survival from ASO of children with first PAH detection within 1 year after ASO was worse than survival of those with first PAH detection more than 1 year after ASO (p=0.039, figure 2A). However, survival from first PAH detection did not differ between these two groups (p=0.409, figure 2B).

{kind=link}
{kind=link}
Potts shunt- and transplant-free survival of children with first PAH detection within and more than 1 year after ASO. (A) Survival from ASO. Survival of 1, 3, 5 and 10 years was 100%, 74%, 59% and 40% for the 18 children with first PAH detection within 1 year after ASO and 100%, 100%, 100% and 100% for the 7 children with first PAH detection more than 1 year after ASO, respectively (p=0.039). (B) Survival from first PAH detection. Survival of 1, 3, 5 and 10 years was 100%, 75%, 60% and 45% for the 18 children with first PAH detection within 1 year after ASO and 100%, 71%, 57% and 57% for the 7 children with first PAH detection more than 1 year after ASO, respectively (p=0.409). ASO, arterial switch operation; PAH, pulmonary arterial hypertension.
During follow-up, four children underwent atrial balloon septostomy for treatment of PAH. Fourteen children (50%) received supportive therapies, including anticoagulation, diuretics and/or oxygen treatment. One child was on calcium channel blocker monotherapy after the parents had refused intravenous epoprostenol therapy, while other PAH-targeted therapies were not available at that time. Six children (24%) received PAH-targeted monotherapy, eight (32%) dual and ten (40%) triple therapy (table 3).
Of the 11 children that did not die, neither underwent lung transplantation nor received a Potts shunt during follow-up; three children were in World Health Organization functional class (WHO-FC) I, seven in WHO-FC II and one in WHO-FC III at last follow-up (median time from first PAH detection, 4.4 years (IQR 1.8, 5.1)).
Discussion
The current study describes a cohort of 25 children with a specific association: children with TGA that developed severe, progressive PAH after timely and successful ASO and in the absence of haemodynamically relevant residual lesions. PAH could present very early after ASO and after several years. Despite the intense use of PAH-targeted therapies, Potts shunt- and transplant-free survival in this cohort was poor with a 5-year survival rate of 73% after ASO and 58% after first PAH detection. In these children, in whom ASO was performed at a median age of 8 days, there was no long-term shunting as trigger for the development of PAH. Therefore, these patients challenge the general concept of PAH associated with CHD, in which prolonged exposure to increased pulmonary blood flow due to a shunt defect is considered the trigger for the development of advanced and irreversible pulmonary vascular remodelling.
Incidence of PAH after neonatal ASO for TGA
For this study, the total number of ASO for TGA performed at each participating centre or within each national cohort during the study period was not available. Therefore, the design of this study precluded the determination of an incidence rate of the association of PAH after successful neonatal ASO for TGA. After conscientiously studying the literature regarding PAH and ASO for TGA, we identified cases of PAH during follow-up in cohorts of children that underwent neonatal ASO for TGA. Out of 100 ASO procedures, Rivenes et al 15 described one child who underwent ASO at the age of 4 days and developed PAH at the age of 42 months. In a series of 156 patients, Losay et al 16 mention one patient with PAH after ASO at the age of 2 weeks. Cordina et al 17 describe a patient who underwent ASO for simple TGA in the neonatal period and presented with PAH at the age of 16 years. Since the latter study included only patients ≥17 years of age, it was not included in incidence estimation. Finally, Roofthooft et al 18 identified one patient with PAH (included in the current study) from a consecutive series of 112 patients who underwent ASO. From these data, it can be roughly estimated that PAH after neonatal ASO for TGA may occur in 0.6%–1.0% of the cases. This estimated incidence, based on literature review, precludes a merely coincidental concurrent occurrence of idiopathic PAH and TGA. The true incidence of the concurrence of TGA and PAH may be even higher, since no systematic screening program for PAH after ASO has been reported, and consequently patients with PAH might have been missed. Furthermore, prenatal or early severe PVD may cause children with TGA to die even before they undergo ASO.19
Clinical characterisation
The male predominance observed in this cohort is in contrast to the generally observed female predominance in paediatric PAH.2 This can be explained by the reported overall male predominance in TGA.20
In this cohort, we identified children with early-onset PAH, that is, first PAH detection within 1 year after ASO, and children with late-onset PAH who had first PAH detection several years after ASO. Survival from ASO was better in this latter group, intrinsically associated with its definition. The observation that survival from first PAH detection did not differ between both groups implies that the PAH in the late-onset group actually developed later in life than in the early-onset group, instead of simply being detected later. Therefore, early-onset and late-onset PAH may represent two different phenotypes, and we hypothesise that both phenotypes may be part of the spectrum of PVD associated with abnormal prenatal haemodynamics in TGA. We were not able to identify discriminating clinical characteristics in the early-onset or late-onset patients.
In the current clinical classification for PH (Nice, France, 2013), PAH associated with CHD is classified based on shunt status.21 Since neonatal ASO for TGA precludes the presence of long-term postnatal shunting, such classification is not suitable. It has been previously advocated that paediatric PVD has specific aspects that are not sufficiently covered in the current classification for PH, including insults on developing and growing organs.3 ,22 ,23 The clinical entity described in this manuscript illustrates the need for further ‘paediatric adaptations’ of the current classification, with special attention for CHD other than shunt lesions and for the concept of programming due to prenatal or postnatal abnormal conditions.
After first PAH detection, prognosis was poor and comparable with that of children with idiopathic PAH in the current era.4 This is in sharp contrast to the excellent long-term survival after ASO reported to be around 98% after 15 years, with approximately 80% freedom from reintervention.11 Thus, this study shows that the occurrence of PAH early or late after ASO represents an important prognostic factor that significantly worsens prognosis. Therefore, the authors advocate that routine lifelong follow-up for children who undergo ASO for TGA should include screening for PAH to allow for early treatment initiation.
Although the proportion of children receiving PAH-targeted dual and especially triple therapy in this study is relatively high compared with what has been previously reported,4 a more aggressive and goal-oriented treatment strategy with early use of PAH-targeted combination therapy, as has been suggested for idiopathic PAH, seems to be justified also in these children.24
Underlying mechanisms
Alterations in prenatal pulmonary haemodynamics have been reported in fetuses with TGA, including restriction or closure of the foramen ovale or ductus arteriosus. It has been suggested that such fetal alterations are associated with altered prenatal flow pattern and mixing pattern resulting in hypoxia of the prenatal pulmonary circulation and increased bronchial circulation.14 ,25 ,26 These alterations may contribute to the development of PVD, already prenatally, in fetuses with TGA. These fetuses are then at risk for rapid postnatal deterioration and death, and also for developing postnatal PH.14 ,19 ,25 ,26 These prenatal haemodynamic alterations may injure or program the developing pulmonary vasculature, leading to abnormal postnatal responses. For the current study, fetal echocardiography data were not available. However, 84% of included children underwent an atrial balloon septostomy procedure in the first days of life. This proportion contrasts with the reported incidence of approximately 40% atrial balloon septostomy procedures in infants with TGA,27 suggesting that incomplete mixing and prenatal haemodynamic changes were more prevalent in these children. Also, the occurrence of PPHN, a condition of disturbed adaptation of the pulmonary vasculature to postnatal life, is more frequent in children with TGA than in the normal population, supporting this concept of altered prenatal pulmonary haemodynamics. PPHN has been suggested to be associated with abnormal pulmonary vascular responses and the development of PAH later in life.2 ,28 In the current study, three children (13%) had PPHN. In a series described by Roofthooft et al,18 14 of 112 infants with TGA presented with PPHN, of which 4 died preoperatively. One of the ten children with PPHN that did undergo ASO developed PAH during follow-up (10%). We hypothesise that the abnormal prenatal haemodynamics in TGA program the pulmonary vasculature, leading to a spectrum of PVD including PPHN and PAH that may develop early (prenatally or perinatally) or later in life.
Another potential explanation could be that a specific genetic make-up in patients with TGA predisposes for the development of PAH. Known PAH-related genes, such as bone morphogenetic protein type II receptor, have been explored for mutations in patients with PAH associated with CHD, but so far no such mutations have been shown in TGA.29 However, very recently, several candidate genes for TGA were identified.30 Two of these genes, ACKR3 (or CXCR7) and NF1, have also been associated with endothelial dysfunction, pulmonary vascular remodelling and the development of PVD/PH.31 ,32
Strengths and limitations
This study is the first to characterise occurrence, presentation and clinical course of PAH in children after neonatal ASO for TGA. As such, this study identifies this specific clinical entity and provides clinically relevant information regarding its clinical features. The data are derived from nine large paediatric cardiology centres, including four European national registries, enhancing epidemiological strength and data quality.
The retrospective nature of the study is associated with inherent limitations. Patients with PAH after ASO might have been missed. Data regarding the total number of ASO performed in the participating centres or national cohorts during the study period were not available, precluding the determination of an incidence rate of PAH after neonatal ASO for TGA from this study. Also, we could not compare children who developed PAH with children who did not, precluding the identification of risk factors for the development of PAH after neonatal ASO for TGA. In the current study, perinatal asphyxia was described in 17% of the children and might have played a confounding role in the observed association of PAH and neonatal ASO for TGA. The current study does not allow identifying underlying mechanisms for the development of PAH in patients with TGA after ASO, and consequently, we can only speculate in this regard and provide supportive clinical data. Further research is needed to confirm the hypotheses regarding abnormal prenatal haemodynamics and genetic susceptibility.
Conclusions
Although PAH after successful neonatal ASO for TGA has been clinically recognised as a specific disease entity, the current study is the first to clinically characterise this association. In this cohort, two phenotypes could be distinguished: early-onset PAH, presenting weeks to months after ASO, and late-onset PAH, presenting years after ASO. In both phenotypes, prognosis is poor, despite the intense use of PAH-targeted therapies, and comparable with paediatric idiopathic PAH. This observational study did not allow for the identification of risk factors for the development of PAH after ASO for TGA. We speculate on the role of altered prenatal pulmonary haemodynamics in TGA, including abnormal flow pattern and mixing pattern associated with prenatal presence of restrictive foramen ovale and ductus arteriosus, and of genetic susceptibility in these children. The data from the current study imply that routine, lifelong follow-up of children who underwent ASO for TGA should include screening for PAH in order to allow for early treatment initiation.
Key messages
What is already known on this subject?
Paediatric pulmonary arterial hypertension (PAH) is a heterogeneous disease, often associated with congenital cardiac shunt defects. Since the 1980s, transposition of the great arteries (TGA) is treated with the neonatal arterial switch operation (ASO). Despite the absence of established risk factors for the development of PAH in children who underwent successful neonatal ASO for TGA, PAH has been reported anecdotally in these children.
What might this study add?
PAH in children after neonatal ASO for TGA occurs more frequently than expected from coincidence and thus represents a specific association. In a worldwide collaboration, we identified 25 children with PAH after successful neonatal ASO for TGA in a 25-year period. Two phenotypes could be distinguished: early-onset PAH presenting weeks to months after ASO and late-onset PAH presenting years after ASO. Despite the intense use of PAH-targeted therapies, prognosis was poor in both phenotypes.
How might this impact on clinical practice?
Given the fatal outcome of children with PAH after neonatal ASO and the varying age of first presentation, lifelong clinical follow-up of children with ASO for TGA should include screening for PAH to allow for early treatment initiation.
Acknowledgments
The authors thank the Spanish Registry (REHIPED) Coordinating Centre, S&H Medical Science Service, for their quality control, logistic and administrative support to gather the data from the Spanish registry collected in this study.
References
Footnotes
Contributors WMHZ and RMFB designed the study, collected data, analysed and interpreted data and drafted the manuscript. OE, SP, DI, DB, PL, ML, JLG, AT-V, AM, MJdC and SM contributed to the conception and design of the study, the data collection or analysis and interpretation of data, revised the draft of the manuscript critically for important intellectual content and finally approved the submitted manuscript.
Funding This research was supported by the Sebald Fund, The Frederick and Margaret L Weyerhaeuser Foundation, The Jayden de Luca Foundation and the Association pour la Recherche en Cardiologie du Foetus à l'Adulte. The REHIPED Registry is supported by unrestricted educational grants from Actelion, Ferrer, GlaxoSmithKline and Pfizer.
Competing interests SM has acted as a consultant to Actelion Pharmaceuticals and Bayer HealthCare. DB has acted as a consultant to Actelion Pharmaceuticals, Novartis, Pfizer, Lilly and Bayer HealthCare. The University of Colorado contracts with Actelion, Bayer, Gilead, Lilly, Pfizer and United Therapeutics for consultancy activities of DDI. The University Medical Center Groningen contracts with Actelion, Bayer, GlaxoSmithKline, Lilly and Pfizer for advisory board and steering committee activities of RMFB. All other authors have nothing to disclose.
Ethics approval Institutional Review Boards of the constituent/participating registries.
Provenance and peer review Not commissioned; externally peer reviewed.