Article Text

Statistics from Altmetric.com
Patients with a bicuspid aortic valve (BAV) may present with aortic regurgitation requiring valve replacement in young adulthood and a subset of patients manifests with progressive aortic dilation and an increased risk of aortic dissection. However, most patients with a BAV develop significant valve dysfunction only very late in life with aortic stenosis due to superimposed fibrocalcific changes. This late phenotypic presentation has hampered our understanding of possible inheritance patterns of this common congenital condition.
In this issue of Heart, Galian-Gay et al 1 found a BAV on echocardiographic screening in 6.5% of 724 first-degree relatives of 256 BAV patients, consistent with significant heritability although valve morphology was not concordant within families. In addition, 9.6% of relatives with a trileaflet valve were found to have aortic dilation, defined as 1.96 SD greater than expected for age, body size and sex (figure 1). Aortic dilation was more prevalent in patients with arterial hypertension (OR 4.48; CI 95% 2.51 to 7.99; p=0.0001) and valvular regurgitation (OR 5.87, CI 95% 1.37 to 25.16; p=0.025).

First-degree relative of a patient with BAV showing a tricuspid leaflet valve in diastole (A) and systole (B) with ascending aortic dilation (C) in the bidimensional echocardiographic images. CT showed a mini-raphe (arrow) between left and right coronary cusps (D) and confirmed ascending aortic dilation (E). Four-dimensional flow MRI revealed the eccentric direction of the aortic jet towards the anterior wall of the ascending aorta (F). BAV, bicuspid aortic valve.
In an editorial, Capoulade et al 2 discuss the clinical implications for families of patients with a BAV (figure 2). They suggest echocardiographic screening of first-degree relatives is appropriate because of the higher prevalence of aortic dilation, although current guidelines do not consistently make this recommendation. Interestingly, further imaging often reveals partial fusion of the aortic valve leaflets or a prodromal BAV in relatives with aortic dilation. Even so, ‘the number of patients with a clinically significant aorta dilation defined by aorta >45 mm is very low (0.4% in first degree relatives with trileaflet aortic valves) and the progression of a ‘minimal’ dilation could also be insignificant in the future, without any impact on management and morbidity/mortality. Further large clinical studies should be designed to address these issues.’

Aortic valve phenotype and related abnormalities in first-degree relatives (FDR) of patients with bicuspid aortic valve (BAV). Tricuspid or BAV phenotypes in FDR of patients with BAV are associated with different risks of developing BAV-related complications, which further impact their follow-up and management. A prodromal (or non-diagnostic) form of BAV, defined by the presence of a mini-raphe, seems at least for some patients associated with BAV-related complications, supporting that this phenotype should be considered as BAV expression and then that these patients will benefit of a closer follow-up as it is suggested for relatives with BAV. Occurrence of BAV phenotype in FDRs of sporadic BAV is approximately 6%–7%, but the prevalence of the prodromal BAV form remain to be determined. TAV, Trifleaflet aortic valve.
Other interesting papers in this issue include a study by Chapman et al 3 that compared three approaches for risk stratification in in 1920 patients with a suspected non-ST elevation acute coronary syndrome. The primary outcome of myocardial infarction or cardiac death occurred in 14% at 30 days. The three diagnostic approaches were similar in excluding an adverse outcome in about 2/3 of patients. In addition, negative predictive values (NPV) were high both for a low cardiac troponin level using a novel assay (NPV 99.5%) and for the European Society of Cardiology (ESC) 1-hour pathway (NPV 99.0%) (figure 3). However, the European Society of Cardiology (ESC) 3-hour pathway, which relies on a diagnostic troponin threshold of 99%, missed 25 events (in contrast to five events with the other approaches) with an NPV of 98.0%.
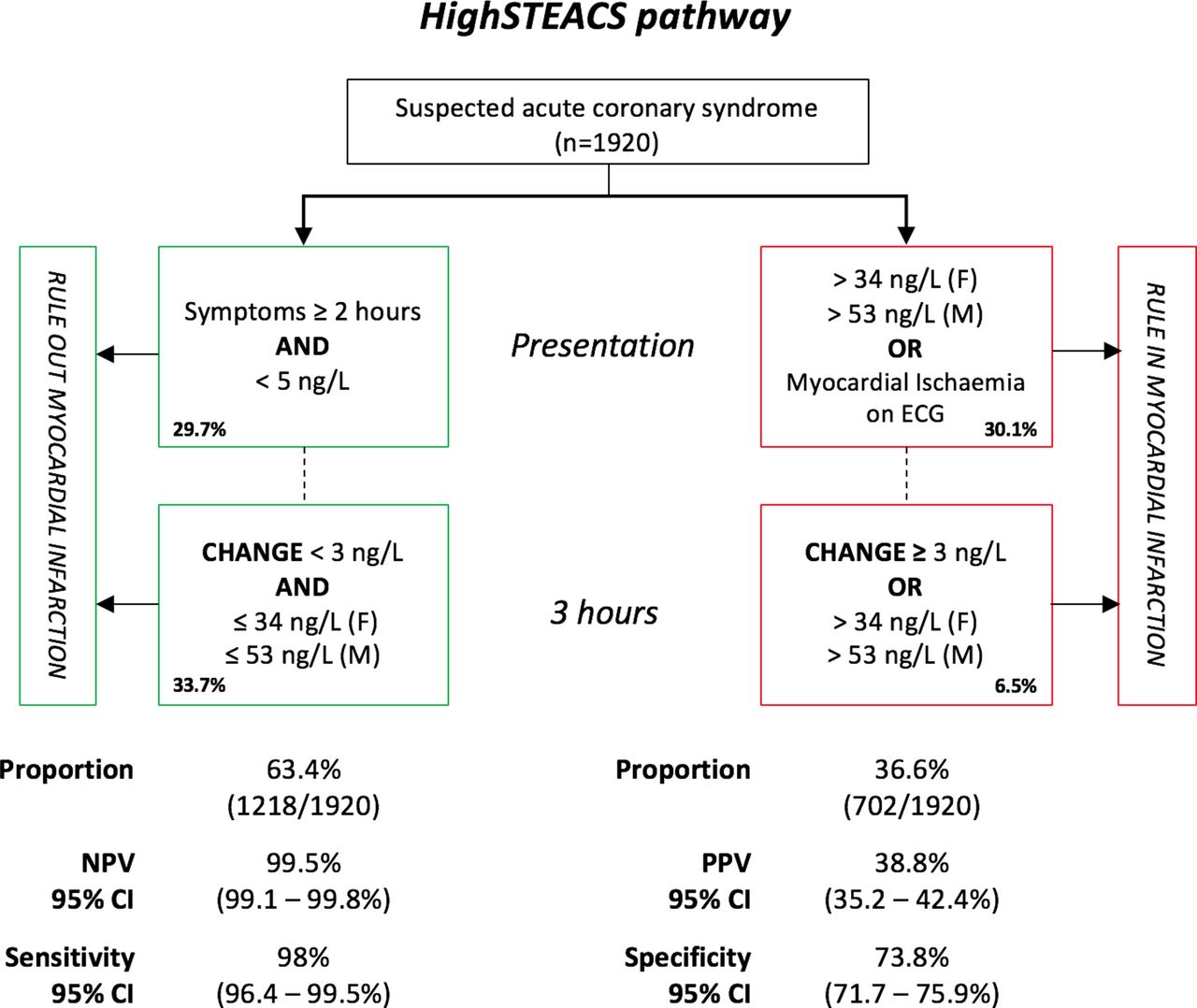
Diagnostic performance of the High-Sensitivity Troponin in the Evaluation of patients with Acute Coronary Syndrome (High-STEACS) pathway. NPV, negative predictive value; PPV, positive predictive value.
In the accompanying editorial, Neumann and Westermann4 discuss the challenges in interpreting troponin levels given the number of commercial assays available, each with different limits of variation and normal ranges. However, as the current study shows, strategies based on low cut-off concentrations will miss fewer patients at risk of adverse outcomes, compared with the traditional approach using blood levels over the 99th percentile. As the authors so succinctly state: ‘The main question is: which patients do we aim to identify? Those with an acute MI requiring urgent revascularisation or those with myocardial injury, who are at high risk and require more careful evaluation. Troponin concentrations are influenced by numerous confounders, such as sex, age,kidney function and many more. Therefore, in future, a more individualised approach including troponin concentrations, their dynamic change, but also adjustment for confounders might improve our understanding of elevated troponin concentrations.’
Both an original research article5 and an editorial6 in this issue address the increasing use of heart or heart-lung transplantation for patients with congenital heart disease (CHD). Compared with other types of end-stage heart disease, those with CHD in the UK are less likely to receive a heart transplant or mechanical circulatory support. There are several barriers to treatment of advanced CHD including limited donor numbers, anatomic complexity, variability in patient management and few cardiologists with expertise in caring for these patients. The authors suggest a need to develop multidisciplinary teams to address the expanding population of adults with CHD.
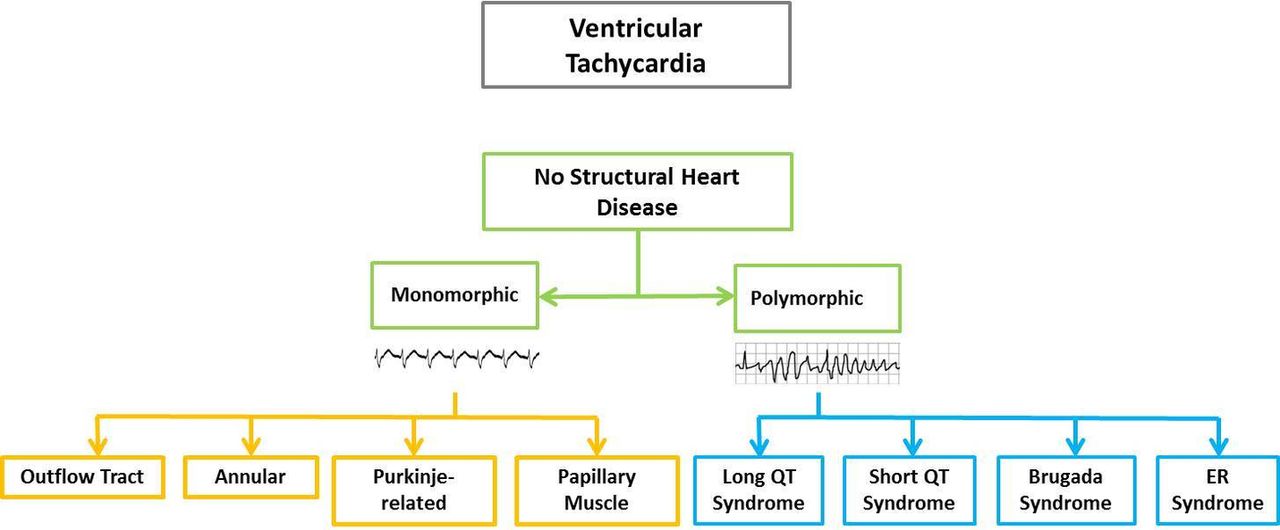
The Education in Heart article in this issue7 discusses the mechanisms and types of ventricular tachycardia (VT) occurring in the absence of structural heart disease (figure 4). The three main mechanisms are re-entry (such as scar-mediated VT), triggered activity (such as right ventricular outflow VT) and automaticity (such as idiopathic focal VT) with clinical presentations varying from palpitations to sudden cardiac death. The risks associated with each type of VT and the options for treatment, including catheter ablation, are reviewed.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Classification of ventricular tachycardia in the absence of structural heart disease.
The Cardiology in Focus article by Dacre8 provides a short history of women in medicine in Britain with the conclusion: ‘Women have come a long way in medicine, but true equality has not been achieved yet. Recognition that a problem exists is the first step towards resolving it. Cardiology is one of the specialties with the smallest number of women, but, with other specialties, is making some progress.’ It is time for all of us to help recognise and seek productive solutions to increasing equity for both cardiovascular disease specialists and patients in the UK and the world.
Footnotes
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.
Patient consent for publication Not required.