Article Text

Abstract
It is estimated that half of all patients with heart failure (HF) have HF with preserved ejection fraction (HFpEF). Yet this form of HF remains a diagnostic and therapeutic challenge. Differentiating HFpEF from other causes of dyspnoea may require advanced diagnostic methods, such as exercise echocardiography, invasive haemodynamics and investigations for ‘HFpEF mimickers’. While the classification of HF has relied heavily on cut-points in left ventricular ejection fraction (LVEF), recent evidence points towards a gradual shift in underlying mechanisms, phenotypes and response to therapies as LVEF increases. For example, among patients with HF, the proportion of hospitalisations and deaths due to cardiac causes decreases as LVEF increases. Medication classes that are efficacious in HF with reduced ejection fraction (HFrEF) have been less so at higher LVEF ranges, decreasing the risk of HF hospitalisation but not cardiovascular or all-cause death in HFpEF. These observations reflect the burden of non-cardiac comorbidities as LVEF increases and highlight the complex pathophysiological mechanisms, both cardiac and non-cardiac, underpinning HFpEF. Treatment with sodium-glucose cotransporter 2 inhibitors reduces the risk of composite cardiovascular events, driven by a reduction in HF hospitalisations; renin-angiotensin-aldosterone blockers and angiotensin-neprilysin inhibitors result in smaller reductions in HF hospitalisations among patients with HFpEF. Comprehensive management of HFpEF includes exercise as well as treatment of risk factors and comorbidities. Classification based on phenotypes may facilitate a more targeted approach to treatment than LVEF categorisation, which sets arbitrary cut-points when LVEF is a continuum. This narrative review summarises the pathophysiology, diagnosis, classification and management of patients with HFpEF.
- heart failure
- diastolic
- pharmacology
- clinical
- diagnostic imaging
- outcome assessment
- health care
Statistics from Altmetric.com
Recent concepts in diagnosis of HFpEF
The evolving definition of HFpEF
Almost two decades ago, it was demonstrated that patients with heart failure (HF) and mild or no reduction in left ventricular ejection fraction (LVEF) had better outcomes than patients with severe systolic dysfunction.1 Mechanistic studies revealed that some of these patients even had normal filling pressures at rest and that a complex interplay existed between pathological cardiac and non-cardiac processes.2 Heart failure with preserved ejection fraction (HFpEF) was first defined as HF in patients with an LVEF >40%.3 Major cardiovascular societies then introduced HF with mid-range and subsequently, mildly reduced ejection fraction (HFmrEF), to describe HF with LVEF 40%–50%.3 4 HFpEF is now defined as HF with LVEF >50%, in absence of prior reduced LVEF.4 However, these classifications of HF according to arbitrary cut-points in LVEF do not appear consistent with recent evidence, which points to a gradual shift and considerable overlap in underlying mechanisms, phenotypes and response to therapy as LVEF increases (figure 1).5–8 HF therapies have generally not been effective at reducing cardiovascular (CV) death beyond LVEF 40%–50% or HF hospitalisation beyond 55%–60%, reflecting the contribution of non-cardiac comorbidities as LVEF increases.7 8 Meta-analyses and mechanistic studies demonstrated the overlapping pathophysiology and response to therapies between HF with reduced ejection fraction (HFrEF) and HFmrEF.7 8 Thus, the definition of HFpEF will continue to evolve as new information regarding phenotypes emerges.

Evolution of pathophysiological understanding of HFpEF. (A) Old concept of HFpEF as a hypertrophied heart with diastolic failure that evolves into systolic failure over time. (B) Prevailing concept of HFpEF and HFrEF as separate diseases, HFpEF caused by microvascular inflammation and HFrEF caused by cardiomyocyte loss. (C) Emerging concept of heart failure as phenotypes overlapping across the spectrum of LV systolic function. There is a gradual change in underlying pathophysiology, mode of death and response to HF therapies across the LVEF spectrum, with influences from genetics, sex, comorbidities, and lifestyle. CV, cardiovascular; GLS, global longitudinal strain; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; LV, left ventricle; LVEF, left ventricular ejection fraction; NO, nitric oxide; ROS, reactive oxygen species; SV, stroke volume.
Current diagnostic approach and its limitations
HFpEF can be defined as HF with normal LVEF and elevated left ventricular (LV) filling pressure at rest or during exercise.9 It is not feasible to subject all patients with HFpEF to an invasive exercise haemodynamic study. Most guidelines define HFpEF clinically as (1) the presence of symptoms and signs of HF; (2) an LVEF ≥50%; (3) careful exclusion of ‘HFpEF mimickers’; and (4) evidence of elevated LV filling pressure or non-invasive correlates (elevated E:e’ ratio, increased left atrial volume, elevated natriuretic peptides (NP)).4 9
The diagnostic criteria for HFpEF are not without limitations. First, the optimal LVEF threshold to define HFpEF is still debated. Several ‘HFpEF’ randomised clinical trials (RCTs) have used lower LVEF cut-offs (40%–45%) to maximise event rates and thus attain statistical power to demonstrate treatment effect. Patients with HFmrEF appear to have similar pathophysiology and treatment response as those with HFrEF, so their inclusion in trials of HFpEF favours treatments effective in HFrEF.7 8 Second, NP levels may not always guide diagnosis as they tend to be lower in HFpEF than HFrEF, likely due to lower diastolic wall stress and higher prevalence of obesity.9 Third, while these criteria rely on measurements at rest, left-sided filling pressures increase only with exercise in many patients with HFpEF9; patients who do not experience dyspnoea or demonstrate signs of elevated LV filling pressure at rest require advanced testing such as exercise echocardiography or invasive exercise haemodynamics to unmask abnormal diastolic reserve (figure 2).9 E:e’ during exercise has been shown to correlate reasonably well with invasively measured pulmonary capillary wedge pressure (PCWP) (figure 2).10 However, imaging studies during exercise are subject to limitations posed by body habitus and operator experience. Invasive haemodynamics provide a direct measurement of PCWP during exercise, although the diagnostic threshold is still debated (exercise PCWP ≥25 mmHg or ΔPCWP/Δcardiac output slope >2.0 mmHg/L/min).11

LV physiology in normal hearts, early HFpEF and advanced HFpEF. Top panel: example tracings of LV pressure–volume loops at rest (red) and during exercise (blue) illustrating LVEDP (black dots) and EDPVR (black line). Dotted line represents high LVEDP, which causes symptoms of dyspnoea. In a normal LV, during exercise, EDV and ESV are increased, leading to increased stroke volume without increase in LVEDP. In early HFpEF, LV physiology is near normal at rest, but reduced diastolic reserve blunts the increase in stroke volume and increases LVEDP. In advanced HFpEF, the EDPVR is shifted upwards and to the left due to a stiffer ventricle, leading to increased LVEDP even at rest. Bottom panel: example tracings of E:A (pulse wave Doppler) and e’:a’ (tissue Doppler) waves at rest (red) and during exercise (blue). In a normal LV, during exercise E and e’ both increase. In early HFpEF, E is normal or low with low e’ at rest, which can also occur in the absence of HFpEF (eg, with ageing). However, during exercise, e’ does not appropriately increase, leading to a higher E:e’ ratio in HFpEF. In more advanced HFpEF, high left atrial pressure and a stiff ventricle lead to elevated E:e’ ratio at rest. A, mitral valve atrial inflow velocity; a’, mitral annular atrial diastolic velocity; E, mitral valve early inflow velocity; e’, mitral annular early diastolic velocity; EDPVR, end-diastolic pressure–volume relationship; EDV, end-diastolic volume; ESV, end-systolic volume; HFpEF, heart failure with preserved ejection fraction; LV, left ventricle; LVEDP, left ventricular end-diastolic pressure.
Two HFpEF diagnostic algorithms—the H2FPEF score12 and the European Society of Cardiology HFA-PEFF algorithm9—combine clinical characteristics and diagnostic parameters to distinguish HFpEF from non-cardiac dyspnoea (tables 1–2). Implementation of these scores in different populations has demonstrated little overlap between patients with high H2FPEF and HFA-PEFF scores; however, both scores identify patients at high risk of HF events.13 Validation of these scores against invasive haemodynamics demonstrated reasonable performance: the area under the receiver operating characteristic curve was 0.73–0.74.13 Still, up to 23% of patients were misclassified in both scores, typically occurring in patients with low scores who met invasive HFpEF criteria. Thus, low scores may not exclude HFpEF.13 Furthermore, a substantial proportion of patients have an intermediate probability for HFpEF using this classification system and require further testing.4
H2FPEF score
Heart Failure Association PEFF algorithm: stepwise approach in diagnosing heart failure with preserved ejection fraction (P–F)
HFpEF mimickers and specific cardiac aetiologies of HFpEF
While most cases of HFpEF are associated with known risk factors and comorbidities, ‘HFpEF mimickers’ such as lung disease, pulmonary embolism, right-sided HF secondary to pulmonary hypertension and renal failure can present with similar symptoms and signs.9 Also, several specific cardiac disorders can present with HFpEF. These can be classified into diseases that affect the myocardium and those that alter cardiac loading conditions (table 3).9 Cardiac amyloidosis, for example, is present in up to 13% of patients with HFpEF on routine biopsy, even when not clinically suspected.14 Targeted therapies exist for most of these conditions, and it is important that they are excluded when a clinical diagnosis of HFpEF is made.9
Specific cardiac aetiologies underlying HFpEF: overview of specific aetiological diagnoses presenting with signs and symptoms of HFpEF
Recent insights in HFpEF pathophysiology
HFpEF results from a complex interplay between risk factors, comorbidities and cardiac pathology that impact LV structure, haemodynamics and systemic organ function (figure 3). In a normal LV, volume increases during diastole are accompanied by minimal pressure increases due to enhanced diastolic suction; LV end-diastolic pressure (LVEDP) remains normal with exercise. In HFpEF, due to increased chamber stiffness, the diastolic pressure–volume relationship is shifted upwards and left compared with a normal LV (figure 2).11 Volume changes thus lead to larger increases in LVEDP. Additionally, in most patients with HFpEF, the exercise-induced increase in cardiac output is blunted due to poor contractile reserve and chronotropic incompetence.10 11 In overt HFpEF, the elevated LVEDP persists at rest, resulting in poor exercise tolerance (figure 2).
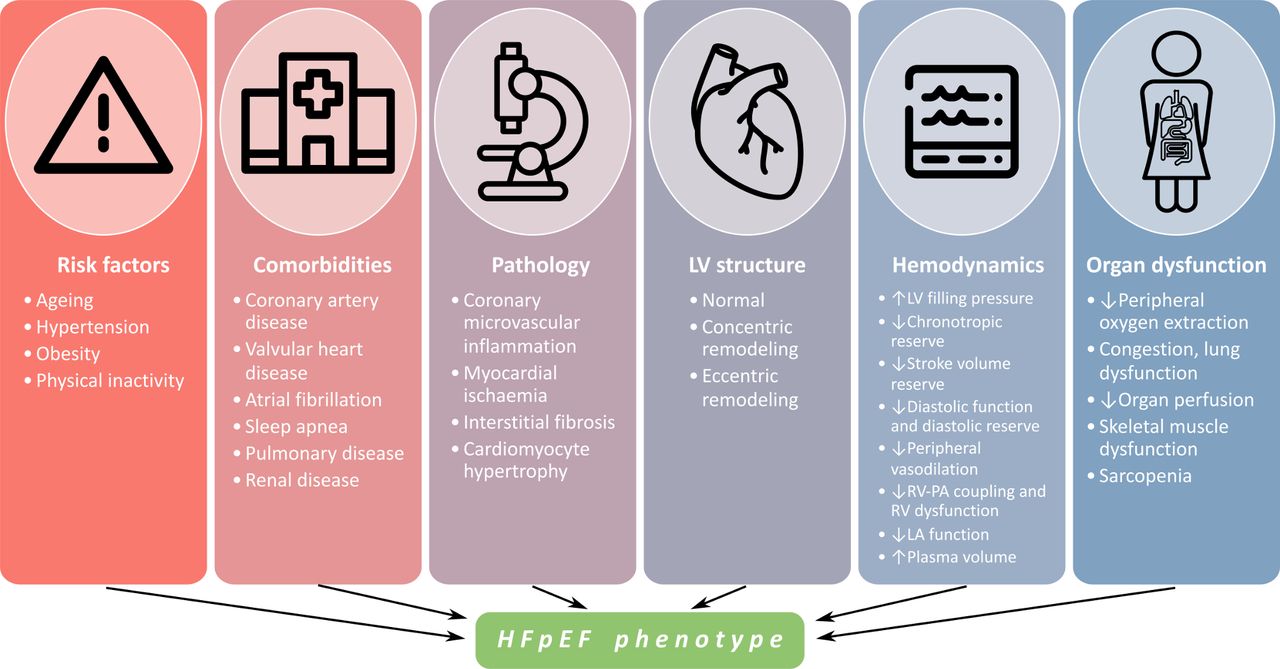
Features of HFpEF. Phenotype is influenced by different risk factors, comorbidities, pathology, LV structure, haemodynamics and organ dysfunction in each patient. HFpEF, heart failure with preserved ejection fraction; LA, left atrium; LV, left ventricle; PA, pulmonary artery; RV, right ventricle.
Cardiac and non-cardiac mechanisms
The importance of non-cardiac mechanisms in HFpEF is highlighted by the decreasing proportion of HF hospitalisations and CV deaths as LVEF increases.1 Non-cardiac abnormalities in HFpEF can be grouped by organ systems.2 Chronic obstructive pulmonary disease, sleep disordered breathing and lung parenchymal disease can result in pulmonary hypertension, eventually leading to right ventricular failure.2 5 Anaemia is a common comorbidity, contributing to exercise intolerance and increased mortality.2 Peripheral vascular dysfunction is frequent and is postulated to play a role in skeletal muscle dysfunction due to impaired oxygen delivery and extraction.2 Chronic kidney disease is present in 50% of patients, and impaired fluid homeostasis in HFpEF is influenced by renal dysfunction. Finally, obesity is both a common comorbidity and a risk factor for developing HFpEF. Regional variations in fat accumulation are associated with different HFpEF risk profiles, whereby higher epicardial and visceral fat have the strongest association with HFpEF.15 Plasma volume expansion can further dysregulate fluid homeostasis in obese patients with HFpEF.15 While the complex interaction of all the proposed non-cardiac mechanisms remains unclear, they appear to shift from mechanisms in HFrEF at higher ranges of LVEF, with a gradual change in phenotype and a gradual decrease in the proportion of CV causes of hospitalisation and death as LVEF increases.2 6 Nevertheless, sudden cardiac death accounts for 25% of mortality in HFpEF and may be a therapeutic target. A recently validated risk score can predict patients with HFpEF who are at risk of sudden cardiac death.16
HFpEF phenotypes
The identification of phenotypes—subgroups with similar clinical and pathophysiological characteristics that are distinct from other subgroups—may allow for the identification of specific HFpEF subgroups more amenable to therapy.17 As LVEF increases, patients with HF are more commonly women, more commonly have hypertension and atrial fibrillation, less commonly have ischaemic heart disease, and have lower NP levels.1 5 Machine learning algorithms have separated patients into phenogroups based on the presence of clinical characteristics, biomarker and imaging profiles.5 18 Although external validation is pending for most studies, the importance of this approach is demonstrated by robust stratification of clinical outcomes according to HF phenotypes.5 18 Different HFpEF phenotypes may reflect different underlying pathophysiology.17 For example, cardiomyocyte calcium homeostasis was markedly abnormal in diabetic and hypertensive HFpEF but not in ischaemic HFpEF.17 Obese patients with HFpEF have markedly different clinical, haemodynamic and molecular changes compared with non-obese patients with HFpEF.17 However, current phenogroups are not mutually exclusive and a given patient can fit into different phenogroups, limiting the uptake of this approach.18 Future research should focus on external validation and on integrating phenotype-based classification schemes in clinical practice and in RCT recruitment (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Personalised medical treatment of HFpEF. Different phenotypes (based on clinical, imaging, biomarker and/or transcriptomic data) represented by red, green and blue colours. (A) Conventional approach to HFpEF medical therapy, treating all patients with HFpEF with a one-size-fits-all treatment regardless of phenotype. As a whole, no clinical benefit is observed (less patients improved compared with patients without change or worsened). However, subgroups with benefit may be observed (red phenotype). (B) While a better overall treatment response to SGLT2i led to overall net clinical benefit, still subgroups of patients with better response (blue phenotype) can be observed. (C) Personalised treatment: considering the phenotype-specific response to medical therapy, a targeted approach using specific drugs in specific phenotypes could lead to net clinical benefit for all patients. ARNI, angiotensin receptor neprilysin inhibitor; HFpEF, heart failure with preserved ejection fraction; MRA, mineralocorticoid receptor antagonist; SGLT2i, sodium-glucose cotransporter 2 inhibitor.
Recent developments in HFpEF management
Lifestyle-based therapy
Up to 80% of patients with HFpEF are overweight, and weight loss has beneficial effects on cardiac relaxation and metabolic profile in older patients without HF. In a small RCT in obese patients with HFpEF, a calorie-restricted diet alone or in combination with exercise training (ET) was associated with significant weight loss and an increase in absolute peak oxygen consumption (VO2peak).19 Together, diet and exercise had additive effects.19 A low-sodium diet has been associated with favourable haemodynamic changes in HFpEF.20
ET has beneficial effects in HFpEF and associated comorbidities such as atrial fibrillation and coronary artery disease. A meta-analysis of 8 RCTs with 463 patients with HFpEF found that ET improved VO2peak, 6 min walk distance and quality of life scores.21 A recent RCT compared standard moderate continuous training with high-intensity interval training. A significant improvement in the primary outcome of VO2peak after 3 months of in person ET was shown, regardless of training modality.22 However, the benefit in VO2peak was not sustained during a 9-month home-based supervised extension of training, highlighting the importance of supervised ET.22 Supervised cardiac rehabilitation remains underused in HF overall and is not reimbursed for patients with HFpEF in many countries.
Atrial fibrillation is commonly associated with HFpEF and may account for some of the symptoms. A 6-month exercise programme combining supervised and home-based aerobic exercise resulted in a significant decrease in the recurrence of atrial fibrillation and a reduction in symptom severity at 12 months.23 Other studies have shown benefit of risk factor management, including weight loss and ET, on atrial fibrillation symptoms and severity.24 Future studies should focus on how to successfully implement and sustain ET in these patients.
Medical therapy
Given the pathophysiological complexity of HFpEF and the interplay with commonly associated comorbidities, the treatment of HFpEF should begin with evidence-informed management of risk factors and comorbidities.4 To date, no medical therapy has demonstrated a reduction in all-cause or CV death in trials of HFpEF. Despite a lack of robust evidence, diuretics have been the mainstay of HFpEF management and are recommended for relief of symptoms due to volume overload.4
Beta-blockers are often prescribed in HFpEF to treat comorbidities such as coronary artery disease and atrial fibrillation. A meta-analysis of three medium-sized RCTs demonstrated a reduction in all-cause mortality, but a vast majority had LVEF <50%.25 RCTs of beta-blockers in HFpEF are a major unmet need. It is likely that some patients with HFpEF benefit from beta-blockers, while in others beta-blockers can worsen chronotropic incompetence. Additionally, beta-blocker type (vasodilating, such as carvedilol, vs primary rate-controlling, such as metoprolol) may have differential effects across HFpEF phenotypes.
Renin-angiotensin-aldosterone system (RAAS) inhibitors and mineralocorticoid receptor antagonists (MRAs) have an established role in HFrEF, but have been less effective in HFpEF, likely because the RAAS plays a less prominent pathophysiological role as LVEF increases.7 Trials of ACE inhibitors and angiotensin II receptor blockers (ARBs) have failed to show a significant reduction in all-cause or CV death in HFpEF (table 4), but have decreased the risk of HF hospitalisation.7 26 The TOPCAT (Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist) trial of the MRA spironolactone in HFpEF failed to show an overall benefit in the primary composite outcome of CV death or HF hospitalisation. However, in an exploratory analysis including only patients from the Americas, a small benefit on the primary outcome was noticed.27 In both TOPCAT and the TOPCAT-Americas subgroup, spironolactone was associated with a reduced risk of HF hospitalisation. Machine learning analysis of the TOPCAT trial identified a phenotype characterised by obesity, diabetes, renal disease and inflammation that exhibited higher risk of CV events and a better response to spironolactone treatment.28 However, these results have not been validated externally, and only an RCT enriched for certain phenotypes will provide evidence for phenotype-based treatment (figure 4).
Primary results and sex differences in major phase III randomised cardiovascular outcome clinical trials that included patients with HFpEF
The angiotensin receptor neprilysin inhibitor (ARNI) sacubitril-valsartan did not reduce the composite of CV death or total HF hospitalisations in patients with HF and LVEF ≥45% relative to valsartan in the PARAGON-HF (Prospective Comparison of ARNI With ARB on Global Outcomes in HFpEF) trial, but significantly reduced HF hospitalisations.29 Of note, patients with elevated troponin or recent HF hospitalisation were at higher risk of CV events and were more likely to benefit from ARNI.30 31 In addition, benefits were higher in patients previously using MRA.7 32 We can speculate that these HFpEF phenotypes—more likely to be associated with structural heart disease and volume overload—may be more responsive to ARNI treatment.
In the EMPEROR-PRESERVED trial, sodium-glucose cotransporter 2 inhibitor (SGLT2i) empagliflozin reduced the risk of composite CV death or total HF hospitalisation in HF with LVEF >40%.33 The benefit was driven by a reduction in HF hospitalisations. There was no statistically significant treatment-LVEF interaction, but benefits did not extend beyond LVEF of 60% in a subgroup analysis.6 Furthermore, in the SOLOIST-WHF (Effect of Sotagliflozin on Cardiovascular Events in Patients with Type 2 Diabetes Post Worsening Heart Failure) trial, the dual SGLT2 and SGLT1 inhibitor sotagliflozin reduced the primary outcome of CV death and HF hospitalisations in patients with diabetes and worsening HF (both HFrEF and HFpEF), driven by reduced HF hospitalisations.34 It is likely that future updates of HF treatment guidelines will include a recommendation to use SGLT2i in HFpEF.
Sex and race or ethnicity differences
Epidemiological studies and randomised trials demonstrate a female predominance in HFpEF (50%–84%) due to differences in age and risk factors; after adjusting for these differences, men and women are at equal risk of developing HFpEF.35 In the TOPCAT-Americas trial, a post-hoc analysis showed no sex differences in the primary composite outcome, but women had a greater reduction in all-cause mortality with spironolactone compared to men (interaction p=0.02).36 In a prespecified subanalysis of the PARAGON-HF trial, the benefits of sacubitril-valsartan on the primary composite outcome (total HF hospitalisations and CV death) were sustained up to a higher LVEF in women (up to 60%) than in men (up to 45%).37 A meta-analysis of trials of RAAS inhibitors was consistent with this finding; the benefits of candesartan, spironolactone and sacubitril-valsartan were sustained to a higher LVEF in women than in men.7 This may be partially explained by differences in LV remodelling due to ageing, with more concentric remodelling in women, leading to a comparatively higher LVEF in women for any given LV volume.38
Analyses of race or ethnicity differences in HFpEF have been limited by underenrolment of Black, Indigenous and people of colour in clinical trials relative to disease prevalence and inadequate reporting of treatment effect by racial or ethnic subgroups.39 In the USA, Black patients have a lower healthcare utilisation and risk of in-hospital mortality from HF, including HFpEF, but may be faced with higher rate of readmissions due to disparities in access to subspecialty and ambulatory care.40 There is no evidence from TOPCAT or EMPEROR-PRESERVED that there are racial differences in treatment effect of MRAs and SGLT2is, respectively.27 33
Integration of remote monitoring and multidisciplinary technology deployment
With recent expansion of telemedicine encounters, clinician–patient interactions are increasingly supported by digital and device innovations.41 Virtual visits have been associated with better adherence to clinic follow-up.41 Implantable remote pulmonary artery (PA) pressure-guided monitoring for patients with HFpEF, New York Heart Association (NYHA) class III symptoms and a prior hospitalisation was associated with a 46% reduction in HF hospitalisations compared with routine care.41 The GUIDE-HF (Haemodynamic-Guided Management of HF) trial, testing whether this benefit extended to patients with NYHA class II–IV symptoms and without a history of hospitalisation, found that PA pressure-guided management did not reduce the composite endpoint of all-cause mortality and total HF events.42 More RCTs are warranted in this field, including novel approaches such as patient-activated therapy and multisensory device algorithms.
Multidisciplinary care integration and health services in HFpEF
HFpEF remains a diagnostic challenge and is often poorly managed, but clinical pathways and multidisciplinary teams can facilitate better care.43 Dedicated HFpEF clinical programmes, nurse visiting programmes or ‘dyspnea clinics’ have been proposed to streamline diagnosis, management and follow-up of patients, but most admissions in patients with HFpEF are secondary to non-cardiac causes.1 A multidisciplinary approach to treatment targeted at common cardiac and non-cardiac comorbidities may help improve outcomes.17 Additionally, transitional care services after hospital discharge improve patient-reported outcomes and may decrease emergency department visits, particularly in women.44–46
Conclusions
Invasive exercise haemodynamics remain the gold standard to diagnose HFpEF, but are not feasible for all patients. Clinical scores as well as exercise echocardiography aid the clinician in discerning HFpEF from its mimickers and from non-cardiac causes of dyspnoea. The three-category classification of HF by LVEF must be reconsidered in light of emerging evidence showing considerable overlap between HFrEF and HFmrEF in pathophysiology and response to treatment. LVEF is a continuous variable, and response to HF therapies appears to be graded; HF pharmacotherapies, including beta-blockers, RAAS inhibitors, ARNI and SGLT2i, that reduce both CV death and HF hospitalisation at lower LVEFs are less effective at reducing death but continue to reduce HF hospitalisation at higher LVEFs. This points to the non-cardiac mechanisms that underpin or accompany HF and differences in underlying pathophysiology as LVEF increases. Care of HF at the higher range of LVEF should entail investigations to exclude mimickers and strategies to address cardiac and non-cardiac comorbidities. Novel insights into HFpEF pathophysiology, including an understanding of sex-based differences and phenotypes, may help narrow down subgroups of patients who may respond to further personalised treatment.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
Acknowledgments
Parts of figure 1 were provided by Servier Medical Art (http://smart.servier.com) used under CC-BY-3.0 licence.
References
Footnotes
ABG and RK are joint first authors.
Twitter @rachkataria, @kevin_damman, @HFpEF, @hvanspall
Contributors HGCV was invited by the journal to provide this review and assumes responsibility for project supervision. ABG, RK and HGCV contributed to the conception or design of the work. ABG, RK, AS and HGCV drafted the manuscript. FZ, KD, KS and SJS critically revised the manuscript. All authors gave final approval and are accountable for the integrity and accuracy of the work.
Funding The study was funded by the Canadian Institutes of Health Research to HGCV, and the Heart and Stroke Foundation of Canada to HGCV.
Competing interests FZ has received fees for serving on the board of Boston Scientific; consulting fees from Novartis, Takeda, AstraZeneca, Boehringer Ingelheim, GE Healthcare, Relypsa, Servier, Boston Scientific, Bayer, Johnson & Johnson, and Resmed; and speaking fees from Pfizer and AstraZeneca. KD received consultancy fees from Abbott and an investigator-initiated study grant from Boehringer Ingelheim; and is supported by the Netherlands Heart Institute (ICIN) and an ESC Heart Failure Association Research Grant. KS is an advisory board member and consultant for Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Cytokinetics, Janssen, Novartis and Novo Nordisk, and receives honoraria. SJS has received research grants from Actelion, AstraZeneca, Corvia, Novartis and Pfizer, and has received consulting fees from Abbott, Actelion, AstraZeneca, Amgen, Aria CV, Axon Therapies, Bayer, Boehringer Ingelheim, Boston Scientific, Bristol Myers Squibb, Cardiora, CVRx, Cytokinetics, Edwards Lifesciences, Eidos, Eisai, Imara, Impulse Dynamics, Intellia, Ionis, Ironwood, Lilly, Merck, MyoKardia, Novartis, Novo Nordisk, Pfizer, Prothena, Regeneron, Rivus, Sanofi, Shifamed, Tenax, Tenaya and United Therapeutics. The other authors report no conflicts of interest with regard to this manuscript.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Commissioned; externally peer reviewed.