Article Text

Abstract
Objectives To determine whether offering cardiac screening to relatives of patients with left ventricular outflow tract obstructions (LVOTOs) would be justified.
Background LVOTOs have been recognised as a group of congenital heart diseases with ‘high heritability’. One of the LVOTOs, the bicuspid aortic valve, is often asymptomatic, but has become known to be associated with sudden, unexpected cardiac death. However, the need for cardiac screening of first-degree relatives of patients with LVOTO has not been determined owing to the lack of studies in well-defined cohorts of consecutive patients.
Methods The families of a cohort of 249 consecutive paediatric patients with LVOTO were offered genetic counselling. Of 182 consenting index patients, 40 patients (22%) appeared to have associated non-cardiac congenital anomalies (LVOTO-NCA). In the other 142 patients with LVOTO, cardiac screening of 449 first-degree relatives was performed.
Results Cardiac screening disclosed a cardiac anomaly in 34 first-degree relatives (8%). In 23 (68%) of these the cardiac anomaly was a bicuspid aortic valve. Twenty-four of these anomalies were newly detected by our screening programme (71%). These 34 cardiac anomalies were found in the families of 28 index cases (20%).
Conclusions This study shows that of the patients with LVOTO without NCA, 20% had (an) affected first-degree relative(s), frequently with undetected bicuspid aortic valves. These data suggest that cardiac screening of relatives of patients with LVOTO without NCA is justified. This may help prevent sudden, unexpected, cardiac death or life-threatening complications in relatives with undetected bicuspid aortic valves.
- Congenital heart defects
- aortic valve
- aortic coarctation
- hypoplastic left heart syndrome
- genetics
- dissection
- sudden cardiac death, paediatric cardiology
- genetics
- aortic valve disease
Statistics from Altmetric.com
- Congenital heart defects
- aortic valve
- aortic coarctation
- hypoplastic left heart syndrome
- genetics
- dissection
- sudden cardiac death, paediatric cardiology
- genetics
- aortic valve disease
Introduction
Left ventricular outflow tract obstructions (LVOTOs) form a group of congenital heart diseases that are generally considered a genetic entity with a high heritability.1 2 The frequency of cardiac anomalies in first-degree relatives, however, has not been studied in well-defined, consecutive patient cohorts. Therefore, the yield of cardiac screening of relatives cannot be assessed using data of previous studies.
LVOTOs are congenital anomalies of the left chamber, the mitral and/or aortic valve, and/or the ascending aorta. A bicuspid aortic valve (BAV), which occurs in approximately 1% of the population,3 4 is included in this group, although it may not be obstructive. Congenital aortic valve stenosis, often caused by BAV, coarctation of the aorta (COA) and hypoplastic left heart syndrome (HLHS) together occur in 8/10 000 live born children.5 Other, less prevalent LVOTOs include mitral valve stenosis, subvalvular or supravalvular aortic stenosis and interruption of the aortic arch. This group of LVOTOs is considered a genetic entity, because various LVOTO diagnoses may occur within families.1 6
Like other congenital heart defects, LVOTOs may occur in combination with non-cardiac congenital anomalies (NCA).7–12 Relatives of patients with NCA were not included in the cardiac screening programme, because the aetiology and heredity of syndromes with heart defects is likely to be different from non-syndromic heart defects.
In LVOTO without NCA, monogenic or more complex (oligogenic or multifactorial) models of inheritance have been suggested and though several loci and genes associated with LVOTOs have been published, the majority of the genes involved is unknown.6 13–22
The BAV, which is the LVOTO lesion with the highest prevalence but often remains unrecognised, is known to be associated with aortic aneurysm and sudden, unexpected cardiac death.23 COA may present with complications such as premature myocardial infarction, cerebral vascular accidents or aortic dissection. To prevent such complications, early detection of these cardiac anomalies is needed. If an increased occurrence of these cardiac anomalies in first-degree relatives of consecutive patients with LVOTO indicates that these relatives are a high-risk population, cardiac screening of these people may be warranted.
This study aims to describe the occurrence of cardiac anomalies in first-degree relatives of patients with LVOTO, after thoroughly excluding patients with additional NCA. We present clinical data, including echocardiographic screening of first-degree relatives, of a well-defined cohort of consecutive paediatric patients with LVOTO and based on these data we propose a diagnostic strategy for patients with LVOTO and their families.
Patients and methods
A cohort of consecutive paediatric patients (n=249), aged between 0 and 18 years, with LVOTO was seen at the Centre for Congenital Heart Diseases, University Medical Centre Groningen (UMCG) between January 2006 and January 2009. The UMCG is a tertiary referral centre for the northern and eastern part of the Netherlands, an area inhabited by approximately 5 000 000 people, with relatively low rates of immigration. A flow chart of the inclusion is shown in figure 1. The cohort included all paediatric patients with LVOTO younger than 18 years old seen both in the clinical wards and in the outpatient clinic during the study period. Terminations of pregnancy and intrauterine deaths were not included. All patients had a detailed cardiac evaluation by a paediatric cardiologist, including ECG and cardiac ultrasound/Doppler imaging. In patients with combined lesions the primary diagnosis was defined as the most relevant anomaly, so if a BAV and a COA were present, the diagnosis was coarctation. All stenotic, normally functioning, or insufficient bicuspid aortic valves were labelled BAV. HLHS was defined as underdevelopment of the left ventricle and ascending aorta together with anomalies of the mitral and/or aortic valve. The group ‘miscellaneous’ includes mitral valve stenosis without HLHS, subvalvular or supravalvular aortic stenosis and interruption of the aorta. All families were offered genetic counselling.
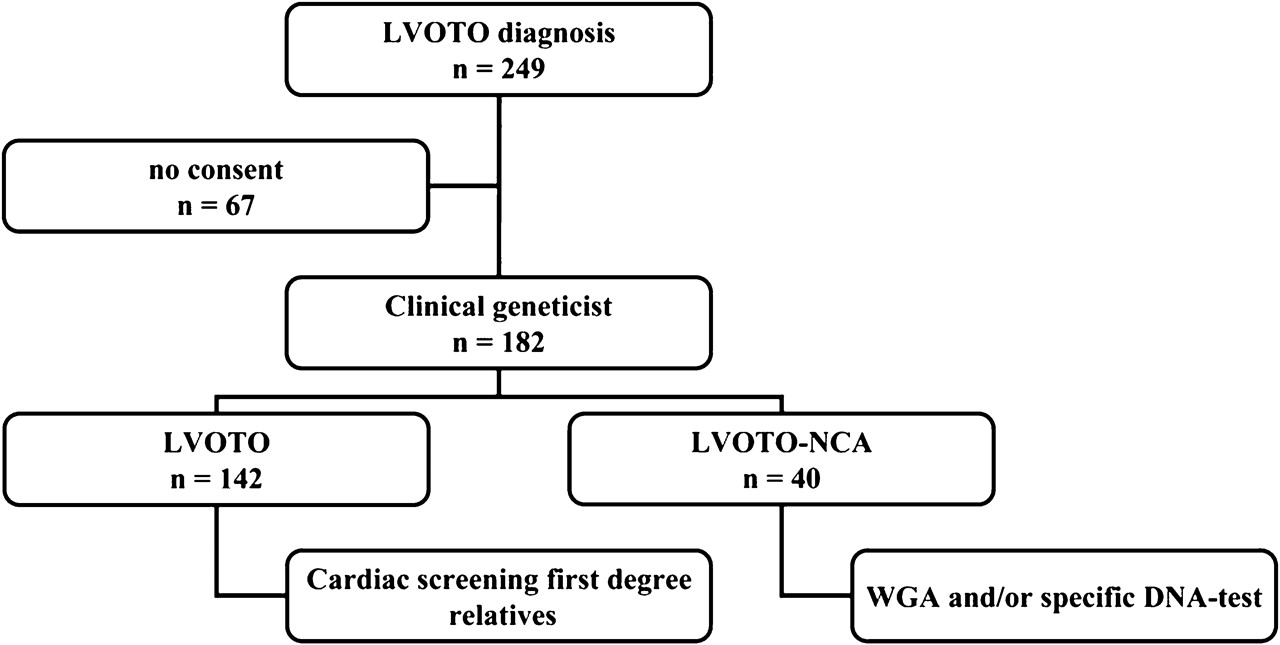
Flowchart inclusion. LVOTO, left ventricular outflow tract obstruction (for definition see text); NCA, non-cardiac congenital anomalies; WGA, whole genome array–comparative genomic hybridisation.
A detailed family history for at least three generations was recorded and, in the case of cardiac anomalies in relatives, these were verified by a written report from the relative's cardiologist. All index patients were evaluated for NCA by a clinical geneticist (WSK-F). Patients with LVOTO-NCA were karyotyped by G-banding. If the karyotype was normal, and no specific diagnosis had been made, array-based comparative genomic hybridisation (whole genome array (WGA)) was performed using a 105 K Agilent oligonucleotide array (custom design ID:019015; Agilent Technologies Inc, Santa Clara, California, USA), according to the manufacturer's protocols. The average resolution was approximately 20 kb. If a deletion was detected, it was confirmed by fluorescent in situ hybridisation (FISH) and the parents were also investigated. If NCA were detected, the relatives were excluded from the cardiac screening protocol, because LVOTO in the context of a de novo chromosomal anomaly or a syndrome was regarded unlikely to occur in asymptomatic relatives of patients with LVOTO-NCA.
All first-degree relatives of patients with LVOTO without NCA were offered cardiac evaluation by a (paediatric) cardiologist, including ECG and a detailed cardiac ultrasound study, including 2D, colour and spectral Doppler imaging, visualising the anatomic components of the left-sided cardiac structures: left atrium, mitral valve, left ventricular cavity, aortic valve, ascending aorta, aortic arch and isthmus. Transverse aortic root dimension was measured at the sinus Valsalva level from the M-mode echocardiogram, at end diastole, from the leading edge of the anterior aortic wall (in accordance with the recommendations of the American Society of Echocardiography).24 If the aortic valve could not be visualised appropriately to judge the separate cusps, additional MRI was performed.
The study was approved by the UMCG ethics committee and all participating families gave their informed consent.
Results
Of the 249 index patients eligible for the study, 182 patients and/or their parents consented to the family investigation (73%). The consenting index patients were 122 male and 60 female subjects (male/female ratio 2:1). NCA were detected in 40 of the 182 patients (22%), 25 male, 15 female. The 142 patients without NCA were 97 male subjects, 45 female subjects, aged 0.5–20.4 years on 1 January 2009 (mean age 8.5 years).
Cardiac diagnosis
The primary diagnosis in 182 index patients was BAV/aortic valve stenosis/aortic insufficiency in 65 patients (45 of these were bicuspid), HLHS in 19 patients, COA (with or without BAV) in 96 patients and miscellaneous in 2 patients. In figure 2 the diagnoses in patients are separated into patients with LVOTO without NCA (figure 2A) and patients with LVOTO-NCA (figure 2B). Diagnoses of patients who did not give consent are also shown (figure 2C). Apart from a relatively low number of HLHS diagnoses in this last group (only one), the distributions are similar.

LVOTO diagnoses in 249 index patients. (A) LVOTO diagnosis in 142 patients with LVOTO; (B) LVOTO diagnosis in 40 patients with LVOTO accompanied by NCA; (C) LVOTO diagnosis in 67 non-consenting patients, who may or may not have accompanying NCA. AVS, aortic valve stenosis; BAV, bicuspid aortic valve; COA, coarctation of the aorta; HLHS, hypoplastic left heart syndrome; LVOTO, left ventricular outflow tract obstruction (for definition see text); NCA, non-cardiac congenital anomalies.
Diagnoses in patients with LVOTO associated with NCA
We found 14 syndrome diagnoses in 40 patients with LVOTO-NCA (35%):
A chromosomal aberration was detected in 11 patients (28%) by karyotyping, FISH or WGA. Of these, five patients had Turner syndrome (caused by a 45,X karyotype in four patients and by a deletion Xp11.23 in one patient). Five patients had a de novo microdeletion in chromosome band 2q24.3q32.1, 3q29, 6p25.3, 15q11.2 and 22q11.2, respectively, and one patient had a mosaic extra ring chromosome 7 (mos 47,XX, r(7)(p22q32)).
A disorder with mendelian inheritance was found in three patients (CHARGE syndrome in two patients, confirmed by mutations in CHD7 and Coffin Siris syndrome in one patient). Clinical details and results of karyotyping, FISH and WGA of these patients are shown in the online supplementary data.
Family history before cardiac screening of first-degree relatives
Before cardiac screening of the first-degree relatives, the three-generation family history was negative for congenital heart defects in 93 families (93/142, 65%), whereas in 49/142 (35%) it was positive for heart defects—namely, LVOTO in 24/142 families (17%) or for congenital heart defects other than LVOTO in 25 families (18%). In 10 of these 49 families the relative previously diagnosed with a heart defect was a parent or sibling.
Cardiac screening of first-degree relatives
Results of cardiac evaluation were abnormal in one or more first-degree relatives in 28/142 families (20%). Cardiac evaluation data were available for 449 of the 483 first-degree relatives (93%): 262/284 parents, (133 fathers, 129 mothers), 187/199 siblings (106 male, 81 female). Reasons for missing data in 34 relatives were: single-parent families, inability to cooperate (in young siblings), ‘not wanting to know’ and missed appointments. Of the 449 first-degree relatives tested, 34 (8%) were diagnosed with a cardiac anomaly (22/262 parents (8%), 12/187 siblings (6%)). The diagnoses of the affected first-degree relatives in these 28 families are listed in table 1. Of the 23 aortic valve anomalies, 20 were bicuspid, 3 tricuspid.
Cardiac diagnoses in first-degree relatives of 142 patients with LVOTO without NCA
In 22 families one first-degree relative was affected, whereas in six families two first-degree relatives were affected. Eight affected siblings of index patients, in six separate families, did not have an affected parent.
In 12 of the 28 families (43%) the three-generation family history was completely negative before the screening, while in 10 families a first-degree relative (five siblings, five parents) was known to be affected, and in six families a further degree relative was affected. The cumulative result of family history and cardiac screening was positive in 61/142 (43%) of the families.
Twenty-four cardiac abnormalities in first-degree relatives (24/34, 71%) were new findings from our cardiac screening programme; the other 10 were already known before this study. One newly diagnosed relative showed dilatation of the aortic root (>40 mm), without a BAV. In one more father an aortic root of 40 mm was detected, which is borderline and will have to be followed up. The cardiac anomalies of the index patients (probands) in these 28 families were found in all LVOTO diagnoses (table 2), though BAV and HLHS showed higher prevalence in relatives than COA.
Affected relatives in 28 affected families
Discussion
In a large cohort of 142 consecutive patients with LVOTO without NCA we showed that in 20% of the index patients a cardiac anomaly was present in one or more of their first-degree relatives. This was most frequently a BAV and in 71% of the affected first-degree relatives the cardiac anomaly had not been previously identified. In 12 (43%) of these 28 multiplex families the three-generation family history before ultrasound screening of the first-degree relatives was completely negative.
NCA were present in 22% of the LVOTO index patients. Eleven patients had numerical chromosomal aberrations (5/11 were Turners' syndrome) and these were likely to be causative because they were all newly arisen in the patients and have been previously described in association with heart defects.7 11 25–30 Details are provided in the online supplementary data. In a previous study on congenital heart disease, a high prevalence of aberrations was also found by array comparative genome hybridisation in selected patients.31 Our data show that a thorough clinical evaluation of NCA in patients with LVOTO is important, since a genetic cause can be detected in 35% of these patients.
In this study we restricted the use of WGA to the syndromic cases. Whether the non-syndromic patients also have chromosomal aberrations we do not know. Therefore, we cannot give an estimation of chromosomal aberrations in all patients with LVOTO. Erdogan et al32 showed that in non-syndromic patients with heart defects chromosomal aberrations may also be detected. However, the occurrence of these chromosomal aberrations is much lower (18/105 (17%)) than the frequency found in our patients with heart defects in combination with NCA (17% vs 35%). Furthermore, whether all these aberrations described by Erdogan are pathogenic is unclear as only three of the 18 were de novo, and only four were previously described as the cause of congenital malformations. Therefore, WGA in non-syndromic patients may reveal new loci for congenital heart diseases in the future, but the yield will be lower than in patients with NCA and the interpretation of pathogenicity will remain a challenge for scientists and clinicians.
The data of this study cannot be compared directly with those of previous studies on the occurrence of cardiac anomalies in relatives of patients with LVOTO because of differences in patient selection and methods. In our opinion, the number of affected families is more relevant than the number of affected relatives, because this reflects the heredity of the disease and bias due to ascertainment of large, affected families is avoided. However, in order to be able to compare our results with previous studies, we adjusted data derived from other studies to the format used in this study, if adequate information was provided. These adjusted data are presented in table 3.
Studies on first-degree relatives of non-syndromic patients with LVOTO
In our study, 34 first-degree relatives (8%) had a cardiac anomaly and 20% of the index patients had an affected first-degree relative. Our results are in contrast with a population-based study39 detecting 13 cases of LVOTO in 1655 first-degree relatives of patients with LVOTO (prevalence 0.79%; RR=12.9). However, this study was not designed to detect asymptomatic cardiac anomalies by cardiac ultrasound. Moreover, information about the number of families was not provided. Recent studies, using cardiac ultrasound screening, show higher prevalences in first-degree relatives than our findings, ranging from 12% to 18% of first-degree relatives and from 37% to 55% of the families. These high numbers, using comparable screening methods including echocardiography, probably confirm the selection bias in these studies due to selection of the most heritable subgroups (BAV and HLHS)33 34 36 38 and, more importantly, to selection towards familial disease in studies with non-consecutive patients.2 33 36–38 Further, most studies did not provide information on the number of previously diagnosed relatives. In the current study, we found the same prevalences as in the study of McBride et al,2 who detected 25 cardiac anomalies in 329 first-degree relatives tested (8%). However, no data were provided that allowed us to calculate the number of affected families. Also no information was provided on how NCA were detected. Therefore, the real prevalence of cardiac anomalies in first-degree relatives of unselected patients with LVOTO without NCA, and with that the yield of cardiac screening of these relatives, cannot be determined, based on previous studies.
BAV was the most prevalent cardiac anomaly seen in asymptomatic relatives in our study, which is consistent with the results of previous studies.2 34–38 As nicely described in a review paper by Braverman, most individuals with a BAV will develop a complication during their life, these complications are often unsuspected and may result in sudden cardiac death.40 Most recent studies focus on the progressive dilatation of the aortic root, which may lead to aneurysm and dissection.41–43 Even in children, dilatation of the aortic root may be progressive: in 50% of the children a size of more than two SDs above the mean is reached 5 years after diagnosis.44 Interestingly, Biner et al found an increased risk of dilatation of the aortic root without a BAV in first-degree relatives of patients with a BAV,45 which is in line with our observation of one relative with dilated aortic root without a BAV and one relative with a borderline value. These studies emphasise that a BAV is not a harmless natural variant, but that it is associated with serious health risks. If patients at risk are identified, aortic dilatation may be postponed by prescription of β blockers44 and sudden unsuspected cardiac death may be prevented by timely surgical intervention.40
The findings of this study, in our opinion suggest that the diagnostic strategy in patients with LVOTO should include: (a) a thorough clinical examination of the patient focused on NCA. If NCA are detected and no syndrome with known mendelian inheritance is recognised, karyotyping, FISH and/or WGA are advised since it reveals abnormalities in 28% of the LVOTO-NCA patients; (b) offering genetic counselling and cardiac screening to first-degree relatives of patients with LVOTO without NCA, because of the high occurrence of cardiac anomalies, often not previously recognised, in those relatives (in 20% of the families). The yield of this strategy in our study population is summarised in figure 3.

{kind=link}
{kind=link}
{kind=link}
Diagnostic strategy for patients with LVOTO. LVOTO, left ventricular outflow tract obstruction (for definition see text); NCA, non-cardiac congenital anomalies; WGA, whole genome array–comparative genomic hybridisation.
The limitations of this study are that we do not have follow-up data on the screened population of first-degree relatives. Therefore, we do not know whether interventions have occurred and whether these have prevented serious complications in this population. The economic impact and cost-effectiveness of our proposed strategy (cardiac screening of an average of three relatives per proband) cannot yet be judged. Another limitation is that our cohort of index patients also contains patients in follow-up and therefore may be under-representing HLHS, owing to the high mortality in this group. If heredity in this most severe group is higher than average, the number of affected families may be underestimated in our cohort. Another limitation of this study is that we do not yet have complete data on NOTCH1 mutation screening in this cohort; this will be part of future research. An obvious limitation of this study, even though we included consecutive patients, is that the possibility of some bias towards syndromic and familial cases, caused by stronger motivation to consent in these families, cannot be ruled out.
We conclude that first-degree relatives of patients with LVOTO have a high prevalence of asymptomatic cardiac anomalies. These are most frequently BAV, and thus relatives of patients with LVOTO carry a risk of serious complications, including sudden cardiac death, which may be preventable. Therefore, offering genetic counselling and cardiac screening including echocardiography, to all families of patients with LVOTO is warranted.
Future research should focus on finding the genes responsible for familial LVOTO, so that all affected relatives can be easily tracked, especially those with asymptomatic BAV who are at risk of preventable sudden cardiac death.
Acknowledgments
We thank Jackie Senior for editing the manuscript.
References
Supplementary materials
Web Only Data for hrt.2010.211433
Files in this Data Supplement:
Web Only Data for hrt.2010.211433
Files in this Data Supplement:
Linked Articles
- Editorial
- PostScript