Article Text

Abstract
Recent advances in cardiovascular magnetic resonance (CMR) now allow the accurate and reproducible measurement of many aspects of cardiac and vascular structure and function, with prognostic data emerging for several key imaging biomarkers. These biomarkers are increasingly used in the evaluation of new drugs, devices and lifestyle modifications for the prevention and treatment of cardiovascular disease. This review outlines a conceptual framework for the application of imaging biomarkers to clinical trials, highlights several important CMR techniques which are in use in randomised studies, and reviews certain aspects of trial design, conduct and interpretation in relation to the use of CMR.
- Clinical trial
- randomised clinical trial
- cardiovascular magnetic resonance
- cardiac magnetic resonance
- MRI
- clinical trials
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Clinical trial
- randomised clinical trial
- cardiovascular magnetic resonance
- cardiac magnetic resonance
- MRI
- clinical trials
Introduction
The randomised controlled trial (RCT) is the gold standard approach to the evaluation of a new proposed therapy for cardiovascular disease.1 2 The growth in the number and scale of RCTs is in part a consequence of the growth in candidate drug targets, driven by new technologies for drug discovery and screening and by a need to test established therapies in new groups of patients.
Cardiovascular magnetic resonance (CMR) offers a range of powerful imaging3 and spectroscopic4 techniques which can be used to identify suitable participants for entry into trials, to ensure baseline comparability of treatment arms and to generate markers of disease presence, severity or activity for use as outcome measures in clinical trials. Many of these CMR-derived markers have been validated,5–14 and increasingly have been shown to be closely linked to important clinical endpoints.15 These developments have led to a rapid increase in their use in clinical trials, a trend which is likely to continue. The versatility of CMR arises from the wide range of sequences available, but only a limited number can reasonably be applied to one individual during a CMR study lasting perhaps 60 min. Each technique differs in terms of its accuracy, precision, reproducibility, sensitivity, link with clinical endpoints and ease of use in terms of both acquisition and analysis. Trialists must therefore understand these characteristics for each outcome measure selected.
The development and evaluation of a potential new therapy is a complex process which varies substantially in terms of time, costs and difficulty, depending on the target condition and the therapy proposed. CMR may have a role in several of the four phases of drug or device development: to identify suitable participants for enrolment and to confirm baseline distribution of key prognostic factors in different treatment arms at any phase of treatment evaluation; to establish efficacy in phase II and III studies, where it may be used to inform a large-scale RCT of ‘hard’ clinical outcomes; to extend the application of a proven therapy to other groups of patients.
Reconciling the expanding pool of candidate therapies and indications for treatment with the desire to have the best possible evidence for efficacy, safety and clinical effectiveness represents a significant challenge.16 One approach is to accept only hard clinical outcome-driven studies, accepting that the resources required to do this mean that only a small number of particularly promising therapies will be evaluated. In this model, CMR may have an important role in identifying the most promising therapies for further evaluation. A second approach is to broaden the scope of therapies but to evaluate them in restricted patient groups, commonly high-risk groups (who are most likely to experience clinical events). Here, CMR may have a role in confirming efficacy in wider patient groups. A third approach has been to measure a circulating biomarker which is linked to disease severity and to assume that, if the biomarker moves in a direction thought to be advantageous, this will correspond to a consequent reduction in clinically meaningful endpoints.17 While such studies may be useful in demonstrating efficacy, recent high-profile studies highlight the risks inherent in assuming that a single circulating biomarker can adequately capture all of the relevant consequences of manipulating a complex biological system.18 For example, in the ILLUMINATE study19 the CETP inhibitor torcetrapib was shown to worsen clinical outcome despite a theoretically favourable direction of movement of circulating biomarkers. CMR and other imaging modalities may have an intermediate role between early studies showing potential benefit in single biomarkers and large-scale RCTs using clinical outcomes. It is of interest that two imaging studies of torcetrapib (RADIANCE 120 and ILLUSTRATE21) did not demonstrate plaque regression. The advantage of imaging surrogate endpoints over circulating surrogate biomarkers is that they capture the downstream activation or common final pathway of disease progression, whereas circulating biomarkers may interrogate a single pathway which may be only one of many relevant pathways. Imaging surrogate endpoint studies can have an important (though not exclusive) role in the evaluation of potential therapies taking place after animal and human dose-ranging studies (phase I–II) but before (and generally as a supplement to or screening process for) large-scale randomised trials. In such cases, it may be that CMR can reliably discriminate clinically important treatment differences with smaller sample sizes than less precise biomarkers, and hence provide results more rapidly and at a potentially lower overall cost than other surrogate endpoints. In all cases, good pharmacovigilance practice will continue to be of great importance.22
Definition of terms
The Biomarkers Definitions Working Group of the US National Institutes of Health defines a biomarker as a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathological processes or pharmacological responses to a therapeutic intervention.23 Surrogate endpoints are those biomarkers which are intended to substitute for clinical endpoints in clinical research studies and, as such, are expected to predict clinical benefit (or harm) based on epidemiological, therapeutic, pathophysiological or other scientific evidence. The distinction between biomarker and surrogate endpoints is important, emphasising that only a proportion of biomarkers can be regarded as surrogate endpoints—namely, those that can predict clinical outcomes. The Working Group definition draws a distinction between biomarker (or surrogate endpoint) for efficacy and biomarker (or surrogate endpoint) for toxicity, usually safety. Imaging endpoints such as those acquired using CMR are predominantly endpoints for efficacy, and there has been a concerted effort within the field in recent years to link imaging biomarkers to clinical outcomes, and these will be augmented by ongoing and future large-scale longitudinal studies.
Surrogate endpoints
The essential feature of a surrogate endpoint is its sensitivity. Clinical events are generally insensitive markers of disease activity because clinical events are generally rare and so typically require large numbers of individuals to be followed for protracted periods. Surrogate endpoints are more sensitive markers of disease presence, severity or activity, are usually continuous variables, and so are ‘common’ and therefore reduce the sample sizes required to have a certain power to detect an effect of given clinical and statistical significance. These reductions in sample size can often be translated into reduced cost and duration of a trial compared with a study based on clinical endpoints or trials using less precise surrogates. The use of surrogate endpoints of all types has come under close scrutiny in recent years. Historically, the CAST trials (in which flecainide and encainide reduced arrhythmias after myocardial infarction but increased mortality) were among the first to highlight the possibility of discordance between the direction of movement of a surrogate endpoint and clinical outcome.24 More recently, the ILLUMINATE study19 showed no net benefit of treatment (indeed, as noted above, it showed potential for harm), yet the biomarker changes would have anticipated benefit. A limitation of all surrogate endpoint studies is that even the best marker of disease presence and activity provides only limited (if any) information about the effects of the drug in question on organ systems or processes which are not the target pathophysiological process. These so-called off-target effects, such as liver dysfunction or QT interval prolongation, are important and need to be rigorously evaluated prior to the universal uptake of such therapies (box 1).
Characteristics of an ideal surrogate imaging endpoint
Accurate
Repeatable
Reproducible
Sensitive
Uniqueness
Prognostically important
Proven interventions lead to change in the surrogate endpoint which translates into improved prognosis
CMR: a range of techniques which can generate imaging surrogate endpoints
In general, CMR as a technique has several strengths which make it, for many applications, the imaging modality of choice for assessing the cardiovascular phenotype. First, it provides inter alia a versatile and sensitive assessment of many aspects of cardiovascular structure and function; these include ventricular function,25 valve function,10 vascular anatomy and function,8 determining the presence and extent of myocardial scar,5 evaluating myocardial perfusion26 27 and myocardial tissue characteristics.28 29 Second, it is safe, well-tolerated, non-invasive and, because no ionising radiation is used (unlike CT and nuclear methods), it lends itself to studies making repeated measures of the same individual—for example, before-and-after treatment phases—with potentially important reductions in sample size (and hence cost) for a given power, statistical significance threshold and predicted treatment effect size. Third, CMR allows assessment of parameters that cannot easily be evaluated in any other way and therefore provides a unique insight into some aspects of cardiovascular disease processes—for example, T2 imaging for myocardial oedema30 and T2* imaging for the assessment of myocardial iron overload.11 31 Fourth, there is now a robust evidence base for the validation and reproducibility of many CMR techniques and an emerging pool of data regarding the prognostic implications of CMR surrogate endpoints.15
Limitations of CMR for clinical trials
Several limitations must be considered when planning CMR clinical trials. Implanted pacemakers, defibrillators and resynchronisation devices generally preclude CMR imaging on safety grounds, although strategies to reduce risk can be employed in the clinical arena.32 This is an important limitation, particularly for studies of patients with advanced cardiac disease in whom treatment effects are perhaps easier to detect and for whom new treatments are most likely to be of benefit.
Another barrier to CMR use in clinical trials is logistics. CMR systems require major investment in hardware, software, infrastructure and maintenance, and access is limited in many parts of the world. The technique is complex and substantial training and experience is required to allow accurate measurements. There may be limited transferability of some sequences across vendors, field strengths and even different sites using the same system and field strength. Standardised protocols for data acquisition and analysis, often with core-lab data analysis, are essential in the clinical trial setting and these have been and are being developed by the Society for CMR, along with a registry of clinical trials involving CMR.33
CMR mandates a period of relative inaccessibility of the participant while data are acquired and so cannot be performed at certain times in a patient's clinical care; this can limit its usefulness in some types of study. An important example is patients with ST elevation myocardial infarction (STEMI) undergoing primary percutaneous coronary intervention (PCI) in whom it is clearly not feasible to perform CMR in the immediate phase of admission prior to urgent reperfusion therapy. This limits the use of before-and-after study designs for the evaluation of new therapeutic adjuncts to primary angioplasty in STEMI. It should be noted, however, that safe imaging of patients with recent myocardial infarction is highly feasible in experienced units with careful safety planning and robust plans for patient evacuation and resuscitation, and has been used in a number of studies.
Optimal time point selection is an important aspect of the application of CMR to clinical trials. Several CMR surrogate endpoints of disease visualise and quantify a highly dynamic process, such as the evolving extent of markers of myocardial reversible and irreversible damage as scar and ventricle remodel in the weeks following a myocardial infarction. This inherent within-patient variability means that time point selection is critical.
The strengths and weaknesses of CMR for clinical trials are shown in box 2.
Strengths and weaknesses of cardiovascular magnetic resonance for clinical trials
Strengths
Safe, non-invasive and repeatable
Versatile
Suitable for statistically powerful repeated measures and crossover study designs
Validation and reproducibility data for many measures
Emerging prognostic data for some measures
Weaknesses
No implanted devices
Complexity and need for specialist training
Limitation for very acute studies (eg, new primary PCI adjuncts)
Dynamic nature of underlying processes (eg, infarct size change) so time point selection important
Lack of prognostic data for some measures
Selected CMR techniques used for clinical trials
A wide variety of CMR sequences, protocols and analysis techniques are available to the trialist. Some of these approaches generate surrogate markers which are widely accepted and some, while validated, remain experimental in a clinical trial context because the prognostic implications of the biomarker are uncertain. The selection of a surrogate marker depends upon the question under study and the way in which a trial will be interpreted. We highlight here just three examples of categories of parameters which can be measured and discuss the extent to which they meet the criteria listed above for a good surrogate marker. We also discuss limitations and give some examples of their use in recent randomised trials.
Vascular imaging
Atherosclerosis is the leading cause of death in developed countries, and emerging therapies will continue to focus on disrupting the pathways which lead to the progression and destabilisation of atherosclerotic plaque. CMR has recently been used in a number of RCTs, and a role complementary to other imaging modalities is emerging for clinical trials in atherosclerosis.34 Quantitative coronary angiography, intravascular ultrasound, optical coherence tomography, carotid-intima media thickness, CT and positron emission tomography all have advantages in different settings, but the strength of CMR lies in its ability to discern distinct plaque components non-invasively, repeatably and with small sample sizes.
Atherosclerosis imaging by CMR can measure a number of aspects of large artery (aorta and carotid) arterial wall structure and function.35 The non-invasive nature of CMR allows serial imaging of the same region of vasculature and the same plaque over time without ionising radiation, although gadolinium-based contrast agents are sometimes needed. Several measures have now been standardised and can be routinely measured by experienced core-labs: wall area and wall volume both measure plaque volume whereas minimal lumen area and plaque eccentricity measure degree of intrusion of the plaque into the lumen and disposition of the plaque, respectively. These measures are highly reproducible36 and agree closely with histological and ex vivo magnetic resonance measures.37 38
Saam et al have provided reproducibility calculations for several CMR atherosclerosis measures based on serial magnetic resonance data from the placebo group of an RCT.39–41 Corti et al used CMR to demonstrate reductions in vessel wall area and vessel thickness in both the carotid and thoracic aorta over 24 months in an RCT of simvastatin in 18 individuals with known atherosclerotic plaque.41 Lee et al 42 showed a decrease in carotid wall area with modified-release nicotinic acid in 17 statin-treated patients over 12 months, several years before large-scale clinical endpoint studies are scheduled to report.
A recent change of emphasis in atherosclerosis imaging is from measures of plaque extent to attempts to evaluate the biological activity or risk posed by a plaque, using techniques which measure plaque composition or biological activity including inflammation or markers of vascular elastic function. CMR can assess the fibrous cap, the lipid-rich/necrotic core, the presence, extent and age of intra-plaque haemorrhage and the relative contribution of loose and dense fibrous tissue to the plaque. AHA lesion classification, soft plaque identification and plaque risk assessment may be achieved.43 The ORION study showed regression of the lipid-rich necrotic core of carotid plaque using rosuvastatin over 2 years,44 demonstrating that serial carotid magnetic resonance could evaluate regression of individual plaque components, with particular significance for a number of emerging proposed therapies for atherosclerosis.
Current research efforts in CMR atherosclerosis imaging are investigating putative markers of the biological activity of the plaque, particularly with respect to inflammation. Ultra-small superparamagnetic iron oxide (USPIO) particles injected intravenously may reflect macrophage activity, and the recent ATHEROMA trial showed a reduction in USPIO accumulation in carotid plaque in subjects randomised to high-dose compared with low-dose atorvastatin.45 CMR can evaluate the functional consequences of atheroma burden on the elastic function of arteries and can measure both regional and global aortic stiffness. Furthermore, the forces and shear stress acting on the wall of the artery can now be measured. Low wall shear stress and high oscillatory shear index have been linked to endothelial activation.
At present, high resolution CMR arterial wall imaging and plaque characterisation is available only for large, relatively immobile arteries, and the walls of the coronary arteries in particular cannot be assessed in this way owing to their small size, mobility and tortuosity. While carotid and abdominal aortic atheroma are of clinical relevance and probably broadly reflect the overall atheroma burden, extrapolation of atheroma regression at one site to other vascular beds may not always be justified. In a study by Yonemura et al the reduction in plaque volume seen in the carotid artery with high-dose atorvastatin was not seen in the abdominal aorta.46
There is correlation between Framingham risk and CMR markers of atherosclerosis,47 and small studies have suggested that carotid CMR findings predict the subsequent risk of stroke,48 49 but the predictive value of such imaging beyond standard risk prediction scores has not yet been proven in large longitudinal studies, although such studies are underway (box 3).
Atherosclerosis imaging by cardiovascular magnetic resonance
Plaque anatomy
Wall area
Wall volume
Plaque lumen effects
Minimal lumen area
Plaque eccentricity
Wall composition
Fibrous cap classification
Lipid-rich/necrotic core
Intra-plaque haemorrhage
Dense/loose fibrous tissue
Wall mechanics
Compliance
Distensibility
Wall shear stress
Oscillatory shear index
Biological activity
Inflammation
Infarct size by late gadolinium enhancement (LGE)
The development of new therapies for the treatment of ongoing or completed myocardial infarction is an important goal in cardiovascular drug and device development. The improvements in mortality and morbidity following myocardial infarction in recent years mean that demonstrating a further reduction requires large sample sizes. A number of CMR techniques are now well established in the clinical evaluation of patients with acute and chronic myocardial infarction and have been reviewed recently.50–52 CMR affords a highly accurate means of quantifying the size5 and transmural extent6 and anatomical location of myocardial infarction (figure 1) and this, combined with accurate measures of left ventricular volumes, systolic function, wall thickness and wall thickening, allows the accurate characterisation of the ‘severity’ of myocardial infarction with much greater sensitivity than clinical endpoints (even when these are combined as composite endpoints). Infarct size is a powerful predictor of subsequent clinical outcome,53 and recent studies have reported that infarct size measured by LGE-CMR is a stronger predictor of clinical outcome than left ventricular ejection fraction or left ventricular volumes.54–56 LGE-CMR may have advantages over nuclear techniques for the evaluation of new therapies in some contexts. Specifically, the increased sensitivity of CMR for subendocardial infarction of <50% transmural extent in both acute57 and chronic infarction58 is likely to reduce sample sizes. Kim et al have recently reviewed the role of CMR in both the clinical management and in infarct size measurement in clinical trials of myocardial infarction59 and make several important points relevant to the use of this technique. First, infarct size evolves over the days and weeks following a myocardial infarction so imaging studies are most likely to be useful after 3 months post-infarct. Second, transparency in reporting the time after the infarct is essential. Third, serial imaging of infarct size is not feasible in many trials of acute myocardial infarction because of the clinical condition of the patient on arrival in hospital and the need for emergency care; thus the advantage of high reproducibility of the technique may be offset by the inability to acquire before-and-after treatment imaging data. As a result, paired tests (before/after) may not be possible and the large reductions in sample size seen in some other contexts25 39 may not be realised. While this is true for the evaluation of therapeutic interventions that need to be instituted within minutes to hours (such as potential improvements of primary PCI), it may be possible to achieve sequential imaging where the treatment is to be administered subacutely (eg, several hours to several days after admission) or with elective procedures (eg, coronary artery bypass graft or complex PCI).60 61 Importantly, LGE used in a parallel (rather than repeated measures) study design still requires only a fraction of the sample size compared with other less reproducible techniques. CMR methods were used to evaluate adjuncts to primary PCI in the CE-MRI sub-study of the EXPIRA study, showing reduced infarct size by LGE-CMR in patients randomised to receive adjunctive thrombectomy using a manual device during primary PCI for STEMI.62
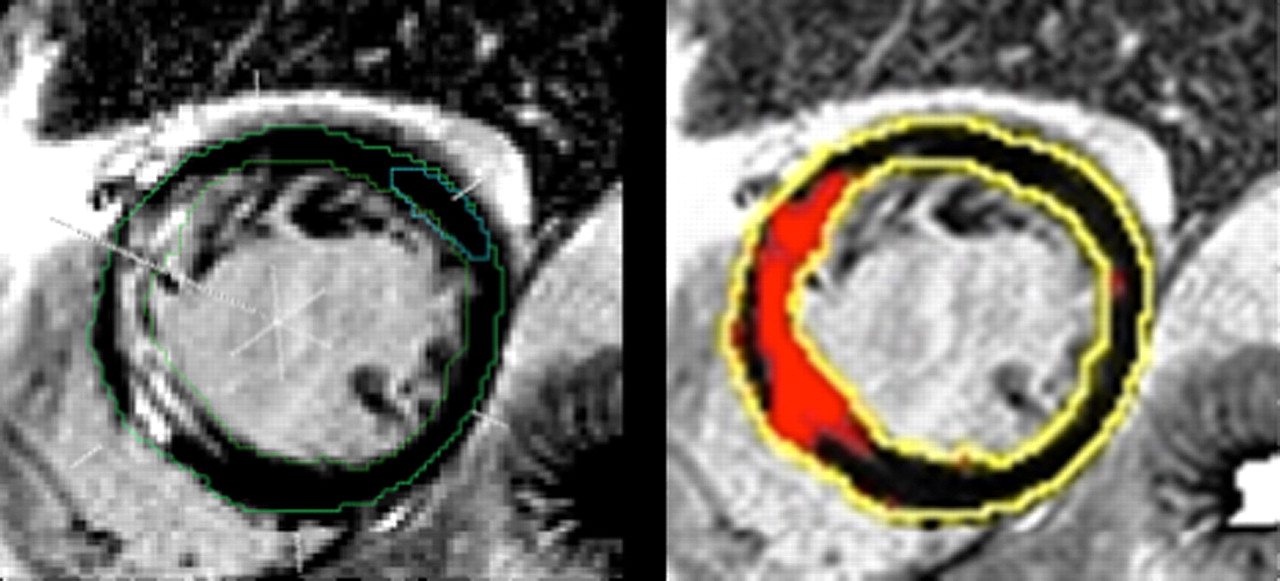
Late gadolinium quantification. Inversion-recovery short axis image of the left ventricle after administration of a gadolinium-based contrast agent showing (A) endocardial and epicardial contours, definition of a region of remote myocardium, and (B) automated segmentation of infarcted from non-infarcted myocardium (based on >2SD pixel intensity compared with remote myocardium).
Ventricular volumes, mass and function
The ability of CMR to measure ventricular volumes, mass and function accurately, reproducibly and on serial studies without significant risk to the patient is a major strength of CMR for clinical trials. Bellenger et al demonstrated substantial sample size reductions (of 81–97%) if CMR measures of volumes, mass and function were serially obtained compared with echocardiography.25 The sample sizes required for left ventricular mass in particular were small (n=9 for each group to detect a 10 g change in left ventricular mass with a power of 90% and p value of <0.05), based on the high reproducibility of this measure by CMR. This is of relevance to conditions in which ventricular performance or responses are important such as hypertension. Several recent studies have evaluated emerging therapies for hypertension using left ventricular mass index as a primary endpoint. The ALLAY study63 used a non-inferiority study design to show that the direct renin inhibitor aliskirin was non-inferior to losartan monotherapy in reducing left ventricular mass index. The effect of the phosphate binding drug sevelamer is currently being evaluated to determine its effects on left ventricular mass in patients with early kidney disease,64 demonstrating that the current use of CMR endpoint studies is not confined to therapies conventionally regarded as being primarily used for cardiovascular indications. CMR may also have a role as part of broader cardiovascular safety studies for drugs intended to treat both cardiovascular and non-cardiovascular conditions.
Current use of CMR in clinical trials
The number of clinical trials using CMR has grown rapidly in recent years. We searched the NIH international trials register for completed registered clinical intervention trials with at least one endpoint including CMR using the search terms (cardiac OR vascular OR cardiovascular) AND magnetic AND resonance.65 We excluded studies which were principally intended to evaluate diagnostic performance of an imaging modality. Fifty-five studies fitted these criteria with total enrolment of 5659 subjects. The mean (SD) sample size was 105 (95) enrolled subjects; 24 studies evaluated treatments for myocardial infarction and nine studies evaluated atherosclerosis. A CMR endpoint was the primary endpoint in 44 of the 55 studies and, in these studies, well-established endpoints such as left ventricular volumes, mass or function were used more frequently (23 studies) than less well-established biomarkers such as USPIO-enhanced MRI signal in carotid plaques (1 study). Unregistered trials and studies registered on other databases were not captured, so these figures probably represent an underestimate of the use of CMR internationally for this type of study.
Clinical trial principles for CMR
The increasing use of CMR for the clinical evaluation of new therapies will require close collaboration between CMR practitioners, pharmacologists, trialists, statisticians and the pharmaceutical industry. Good practice regarding clinical trial design, conduct, analysis and reporting is essential in CMR trials, and training in these areas will be required for CMR practitioners who are involved with clinical trials.
Trial design
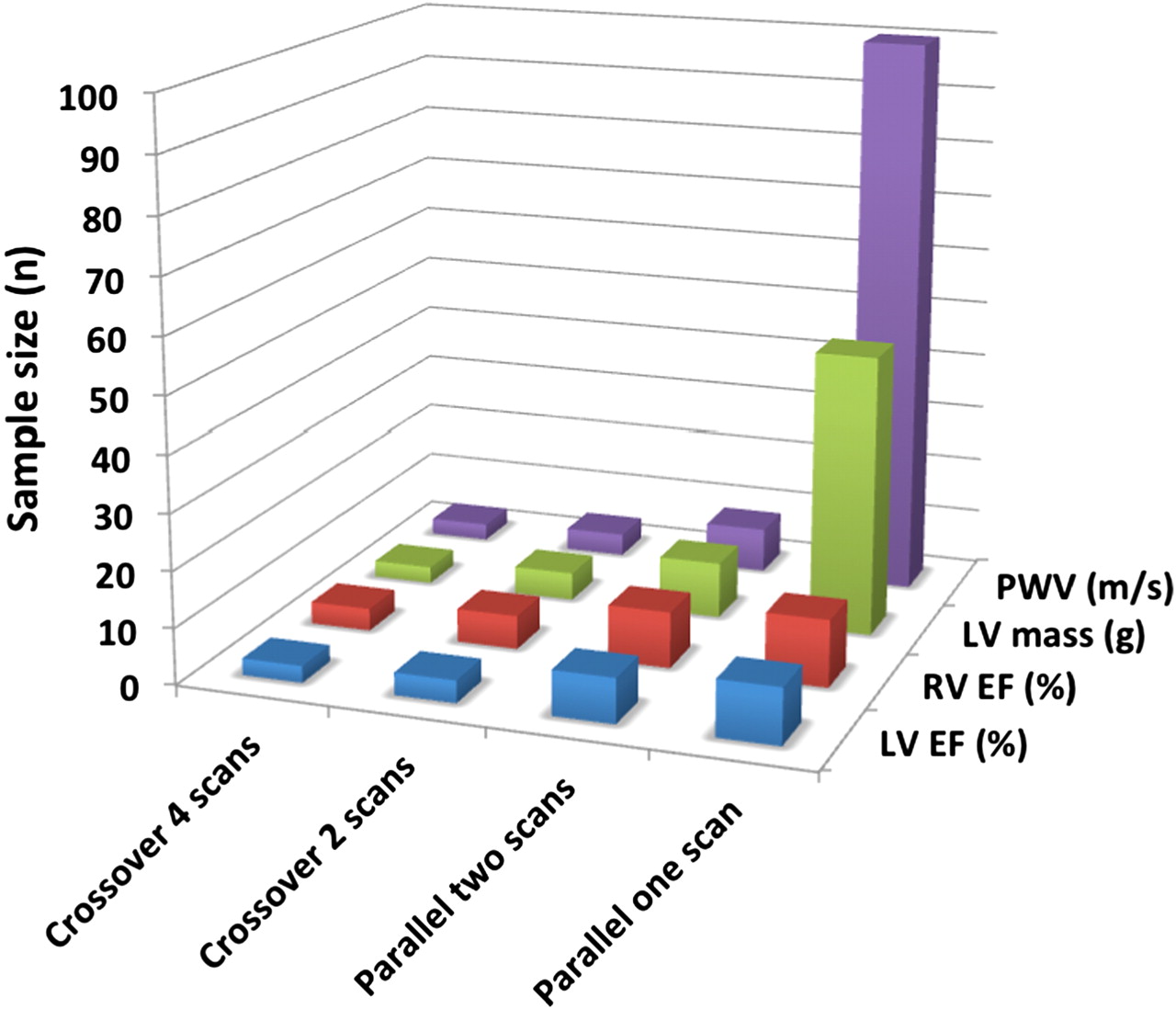
CMR is well suited to a number of study designs (figure 2). Crossover studies, in which each participant is randomly allocated to a sequence of therapeutic arms, are particularly to be encouraged where the therapy in question is likely to have fairly rapidly reversed effects on a long-term condition, in view of the favourable effects of this design on sample size, for a given power and level of statistical and clinical significance (figure 3).

Four different study designs. Upper panels show crossover designs, lower panels show parallel designs. Panels on the left show single cardiovascular magnetic resonance (CMR) scan per treatment phase, panels on the right show before/after scans for each treatment phase.

{kind=link}
{kind=link}
{kind=link}
Effect of study design on sample size, assuming a given level of power and statistical significance, to discriminate a clinically important difference for four cardiovascular magnetic resonance surrogate markers. EF, ejection fraction; LV, left ventricle; PWV, pulse wave velocity; RV, right ventricle.
Trial analysis
CMR sequences, protocols and endpoints should be prespecified in the trial protocol, which should be registered and published.66 One endpoint should be specified as the primary endpoint. CMR outcome assessment can often (but not always) be blinded to treatment allocation. Artefacts related to treatment allocation will sometimes compromise blinding of outcome assessment—for example, artefacts related to sternal wires in trials of coronary artery bypass grafting versus medical therapy.67
Trial reporting
Clinical trials using CMR to evaluate an investigational medicinal product or other intervention should be reported and considered for publication if they address an important clinical question, with robust methods and are adequately powered, regardless of the direction or magnitude of the study findings.68 CMR trials should adhere to CONSORT guidelines for clinical trial reporting,69 and reports should pay particular attention to the method of randomisation, allocation concealment and blinding and should make clear which analyses and endpoints were prespecified (and therefore hypothesis-testing) and which were not (and therefore hypothesis-generating).
Conclusion
More than ever, the process of identifying promising therapies and their evaluation from the laboratory to the clinic requires collaboration between widely differing fields. The versatility and power of CMR to address key issues in the evaluation of therapies for many cardiovascular diseases means that close collaboration will be needed between CMR specialists and others involved in clinical trials. This will ensure best use of the opportunity which CMR affords to evaluate promising new therapies and ultimately to deliver these therapies to patients with cardiovascular disease.
Acknowledgments
The authors would like to thank Dr Alistair Lindsay and Dr Erica Dall'Armellina for helpful comments on the manuscript, and Dr Stefan K Piechnik, Dr Robin Choudhury and Dr Erica Dall'Armellina (funded by the NIHR Biomedical Research Centre at Oxford) for the image showing LGE quantification methods and Dr Stefan K Piechnik for access to the newly developed cardiovascular MR image processing software suite MC_ROI.
References
Footnotes
Funding AP is supported by the British Heart Foundation and the Oxford National Institute of Health Research (NIHR) Biomedical Research Centre. SEP is directly funded by the Barts and the London NIHR Cardiovascular Biomedical Research Unit. DA and PE are NIHR Senior Investigators and acknowledge support from the NIHR Comprehensive Biomedical Research Centre at Imperial College Healthcare NHS Trust.
Competing interests PE is a member of the UK Biobank Steering Committee and Chair of the UK Biobank Enhancements subgroup which is considering addition of CMR measurements in a subsample of UK Biobank participants. AP and SEP are involved in the design and conduct of clinical trials using CMR to evaluate new therapies, some of which involve some funding from the pharmaceutical industry.
Provenance and peer review Not commissioned; externally peer reviewed.