Article Text

Abstract
Background Pulmonary hypertension (PHT) lacks community prevalence and outcome data.
Objective To characterise minimum ‘indicative’ prevalences and mortality data for all forms of PHT in a selected population with an elevated estimated pulmonary artery systolic pressure (ePASP) on echocardiography.
Design Observational cohort study.
Setting Residents of Armadale and the surrounding region in Western Australia (population 165 450) referred to our unit for transthoracic echocardiography between January 2003 and December 2009.
Results Overall, 10 314 individuals (6.2% of the surrounding population) had 15 633 echo studies performed. Of these, 3320 patients (32%) had insufficient TR to ePASP and 936 individuals (9.1%, 95% CI 8.6% to 9.7%) had PHT, defined as, ePASP>40 mm Hg. The minimum ‘indicative’ prevalence for all forms of PHT is 326 cases/100 000 inhabitants of the local population, with left heart disease-associated PHT being the commonest cause (250 cases/100 000). 15 cases of pulmonary arterial hypertension/100 000 inhabitants were identified and an additional 144 individuals (15%) with no identified cause for their PHT. The mean time to death for those with ePASP >40 mm Hg, calculated from the first recorded ePASP, was 4.1 years (95% CI 3.9 to 4.3). PHT increased mortality whatever the underlying cause, but patients with PHT from left heart disease had the worst prognosis and those with idiopathic pulmonary arterial hypertension receiving disease-specific treatment the best prognosis. Risk of death increased with PHT severity: severe pulmonary hypertension shortened the lifespan by an average of 1.1 years compared with mild pulmonary hypertension.
Conclusions In this cohort, PHT was common and deadly. Left heart disease was the most common cause and had the worst prognosis and treated pulmonary arterial hypertension had the best prognosis.
- Pulmonary hypertension
- echocardiography
- prevalence
- cardiopulmonary haemodynamics
- pulmonary arterial hypertension
- PAH
- lung
- pulmonary vascular disease
- imaging and diagnostics
- echocardiography
- heart failure
- arrhythmias
- atrial fibrillation
- st-T alterations
- chronic heart failure
- public health
- epidemiology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Pulmonary hypertension
- echocardiography
- prevalence
- cardiopulmonary haemodynamics
- pulmonary arterial hypertension
- PAH
- lung
- pulmonary vascular disease
- imaging and diagnostics
- echocardiography
- heart failure
- arrhythmias
- atrial fibrillation
- st-T alterations
- chronic heart failure
- public health
- epidemiology
Introduction
Pulmonary hypertension (PHT) is defined as a sustained increase in mean pulmonary arterial pressure (mPAP) to >25 mm Hg, measured using right heart catheterisation (RHC). PHT is typically suspected after echocardiographic investigation of breathlessness; the pulmonary artery systolic pressure (PASP) being estimated from tricuspid regurgitation velocity, using the modified Bernoulli equation. PHT has many underlying causes, including left heart, respiratory and pulmonary vascular diseases. Pulmonary arterial hypertension (PAH) is a subset of PHT, with normal pulmonary artery wedge pressure and increased pulmonary vascular resistance >3 Wood units.1 ,2
PHT has been classified numerous times, most recently via an interdisciplinary collaboration of international societies.2–4 PHT classification accounts for approximately 40 diseases associated with its clinical presentation, characterised by progressive dyspnoea, functional limitation and if untreated, right ventricular failure and death.5 Most PHT-related diseases are not linked and may have potentially different treatments and survival trajectories. However, community-based reports of clinical characteristics, progression and prevalence of specific PHT subtypes are lacking.5–11
We sought to characterise the minimum prevalence and prognostic impact of each PHT subtype in a selected population using a retrospective analysis of individuals with an elevated estimated pulmonary artery systolic pressure (ePASP) on echocardiography.
Methods
This study conforms to the STROBE recommendations for reporting cohort studies.12
Study setting
Armadale and its catchment area (2006 population, 165 450) is located 30 km southeast of the city of Perth, Western Australia. Armadale's population is broadly representative of Australia's population: mean ages being 35 versus 36 years, male population 49.6% versus 49.4%, Australian born 63% versus 70% and indigenous population 2.5% versus 2.6%, respectively. Cardiological management is largely ‘captive’ with a single adult echocardiography laboratory in Armadale managing nearly all local referrals.
Study cohort
We conducted a retrospective echocardiography database search of residents within the Armadale catchment referred to our unit for transthoracic echocardiography between January 2003 and December 2009 and who had an elevated ePASP ≥40 mm Hg. Patients with an ePASP <40 mm Hg or insufficient tricuspid regurgitation preventing PASP estimation were excluded. Mean cohort follow-up was 1060 days (2.9±2.0 years) from the index echocardiogram. The study was approved by the University of Notre Dame Australia human research ethics committee.
Echocardiography
All echocardiograms were obtained by an experienced sonographer using a GE Vivid 7, GE Vivid i (GE Vingmed Ultrasound AS, Horten, Norway), or a Philips iE33 (Philips, Seattle, USA) cardiac ultrasound machine. Images were stored digitally using commercial software (Prosolv Cardiovascular) and reported by a senior echocardiologist in accordance with published guidelines.13 Standard complete two-dimensional echocardiography was performed in all individuals, with chamber quantification assessed according to published criteria.14 PAP was assessed by imaging the peak velocity of the tricuspid regurgitant jet (TRV) and applying the simplified Bernoulli equation (right ventricular systolic pressure (RVSP)=4v2 + right atrial pressure.15 ,16 For standardisation, a right atrial pressure of 10 mm Hg was assumed for all patients unless clear features were present that suggested otherwise. Insufficient tricuspid regurgitation was defined as incomplete TRV or inability to identify peak velocity with confidence.
Identification and coding of pulmonary hypertension
Using the SQL database (Prosolv Cardiovascular echo software), a query search was built. Classified separately, patients with insufficient tricuspid regurgitation did not contribute to PHT prevalence estimates. All patients with an estimated PAP >40 mm Hg between 1 January 2003 and 31 December 2009 were included. Patients with PHT were prospectively separated into three clinically relevant groups: ePASP 40–50 mm Hg (mild), 51–60 mm Hg (moderate) and >60 mm Hg (severe). Patients were coded for referral type and identified PHT causality according to published guidelines.4 ,17 Clinical data and echocardiographic results were used to determine PHT causality (eg, those with a history chronic obstructive pulmonary disease were classified as respiratory-related PHT, unless significant left heart disease was detected). Where required, the patient's general practitioner was consulted for more definitive information. If the cause was still not clear from these data, the cause of the PHT was deemed to be ‘unknown’. In addition, if the findings on echocardiography suggested possible left heart disease, but the degree of PHT appeared ‘out of keeping’ with the degree of left heart disease, the cause was classified as ‘unknown’. In patients with two or more known causes of PHT, the dominant cause was classified.
PHT causality was coded using criteria and subcategories of the ACC working group classifications; group 1: PAH, group; 2: PHT in association with left heart disease; group 3: PHT in association with hypoxic respiratory disease; group 4: PHT due to chronic thrombotic and/or embolic disease and group 5: miscellaneous.18
Two sets of blinded coding were undertaken (including a consultant echocardiologist (DP)) then finalised by consensus or with further consultation with a specialist from respiratory medicine (EG). If causality remained undetermined the case was classified as ‘unknown’.
Identified cases of PAH were managed through the advanced lung disease unit at Royal Perth Hospital. All were rigorously investigated to elucidate the cause of PHT, including respiratory function tests, sleep studies, high-resolution CT scan, VQ scan, CT pulmonary angiogram and RHC.
Study follow-up
Mortality was investigated in all cases from the time of the first recorded ePASP to a census date (31 December 2009). Time to death was established using individual linked data extracted from the Western Australian Department of Health Database.
Statistics
Quantitative variables are described using means±SD and medians with the 25th and 75th IQR where applicable. Cross tabulations using the χ2 statistic were used with non-parametric Kruskal–Wallis test to compare differences. We employed logistical and Cox proportional regression models to evaluate factors associated with mortality (such as age, sex, aetiology of PHT and the degree of PHT). The detection rates of PHT are calculated as the ratio of existing and newly diagnosed patients at the censor date 31 December 2009 (excluding those who had died during the follow-up period) and the estimated total Armadale and surrounding districts adult population according to the last Australian census survey performed in 2006.19 Mortality data are expressed as cumulative survival and with the use of Kaplan–Meier estimates.
Results
Cohort characteristics
Between January 2003 and December 2009, 10 314 individuals (6.2% of the population), had 15 633 echocardiography studies performed for various reasons, including investigations of murmurs, ventricular function, breathlessness and PHT. Of these, 3320 patients (32%) had insufficient tricuspid regurgitation to measure PASP. Nine hundred and thirty-six individuals (9.1%, 95% CI 8.6 to 9.7%) had echocardiographic evidence of PHT (ie, TVR profiles clearly demonstrated the maximum velocity and ePASP was >40 mm Hg). This proportion is slightly greater than the 6.6% reported in the INCIPIT study.20 The characteristics of patients with PHT and included in the analysis are presented in table 1.
Demographics for all patients with echocardiographic evidence of PHT
Of the 936 individuals with elevated ePASP, 636 (68%) patients had left heart disease as the dominant cause, 87 (9%) with respiratory disease (n=74 (7.9%)) or sleep-related hypoventilation (n=13 (1.3%))-associated PHT, 19 (2%) had chronic pulmonary thromboembolic disease, 25 (2.7%) had other diseases (‘miscellaneous’) causing PHT and 144 (15%) had PHT with no known cause from the available data. Thirteen patients had congenital heart disease-associated PAH (CHD/PAH, median age 43 (IQR 39–75), median PASP 56 mm Hg (IQR 50–67)), four patients with scleroderma-associated PAH (SSc/PAH, median age 71 (IQR 62–71), median PASP 94 mm Hg (IQR 80–95)) and eight patients were diagnosed with idiopathic PAH (iPAH, median age 63 (IQR 52–78), median PASP 56 mm Hg (IQR 50–84)). In total, 25 (2.7%) of our final PHT patient cohort had PAH (WHO group 1) confirmed by RHC after thorough evaluation at a tertiary centre. There was borderline significant differences between median ePASP (p=0.0496). All eight patients with iPAH, all four patients with SSc/PAH scleroderma and five of the patients with CHD/PAH received disease-specific PAH treatment during at least part of the study period.
Indicative prevalence of pulmonary hypertension
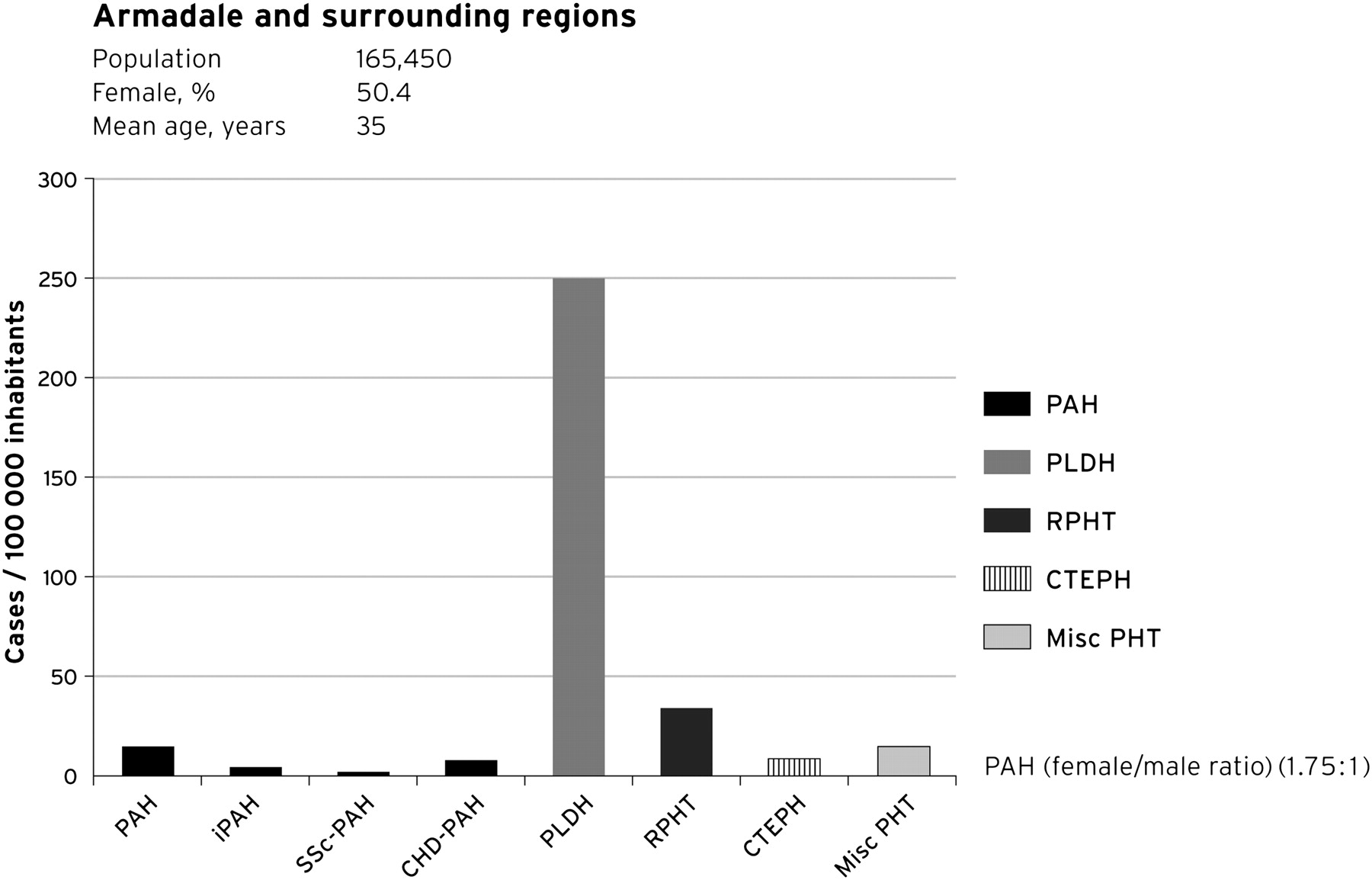
Figure 1 shows the cumulative number of cases (all patients still alive at 31 December 2009) of all forms of PHT in our population, based on ePASP using echocardiography. We calculated a minimum indicative prevalence for all forms of PHT of 326 identified cases/100 000 of the population (groups 1–5). PHT due to left heart disease was the most common cause for PHT in our population (250 cases/100 000), or one in approximately 400 individuals in the community. The range of left heart diseases was broad, most commonly ischaemic and non-ischaemic cardiomyopathies and valvular heart disease.

Cases detected: the ‘indicative’ prevalence of pulmonary hypertension in Armadale and surrounding regions, December 2009. CHD, congenital heart disease; CTEPH, chronic thromboembolic pulmonary hypertension; iPAH, idiopathic PAHs; PAH, pulmonary arterial hypertension; Misc PHT, pulmonary hypertension due to other causes; PLHD, pulmonary hypertension secondary to left heart disease; RPHT, respiratory-associated pulmonary hypertension; SSc, systemic sclerosis.
PHT from respiratory disease affected 37 cases/100 000 population. PAH accounts for a very small percentage of our patients (15 cases/100 000 population). The indicative prevalence of iPAH was 4.8 cases/100 000 population. Patients with PAH were older than the general population, but younger than those with other forms of PHT and there was a female preponderance with PAH as might be expected. In addition, we were unable to code a sufficient cause for the elevated ePASP in 15% (144 cases) of those patients identified through our search. Patients with insufficient TR to estimate RVSP were not included in the calculations.
Survival
Mean survival for patients with ‘all-cause’ PHT (eRVSP >40 mm Hg) patients was 4.3±0.1 years from first recorded echocardiogram (figure 2A). The cumulative survival between each PHT subgroup (WHO groups 1–5) is shown in figure 3A. Patients with pulmonary hypertension caused by respiratory disease fared poorly (mean survival 4.1±0.3 years), similar to those with left heart disease (4.2±0.1 years) and those without an identified cause for their PHT (4.3±0.3 years). By contrast, patients with PAH, most of whom were prescribed disease-specific treatment, had the best survival (82% survival at the conclusion of the study). We did note a significant difference between mortality among the PAH WHO group 1 patients (iPAH 80%, SSc/PAH 40% and congenital heart disease-associated pulmonary hypertension 85% alive at the end of the study of censor, p=0.025). Patients with insufficient tricuspid regurgitation for estimation of RVSP were not included in the analysis.

Kaplan–Meier survival and proportional OR for all causes of pulmonary hypertension. (A) Kaplan–Meier survival for all causes of pulmonary hypertension. (B) Independent variables associated with mortality. PAP, pulmonary artery pressure on echocardiogram in mm Hg.

Kaplan–Meier survival estimates. (A) Each subtype of PHT. (B) Clinical severity of PASP. PAH, pulmonary arterial hypertension; Left heart disease, pulmonary hypertension secondary to left heart disease; Respiratory disease, respiratory-associated pulmonary hypertension; Chronic thromboembolic, chronic thromboembolic pulmonary hypertension; Miscellaneous, miscellaneous pulmonary hypertension; Unknown cause, pulmonary hypertension of unknown cause. Mortality for mild, moderate and severe pulmonary hypertension for all causes of pulmonary hypertension during the period of follow-up. p<0.001 for difference between reference range mild (41–50 mm Hg) compared with moderate or severe. PASP, pulmonary artery systolic pressure.
Associations with prevalent forms of pulmonary hypertension
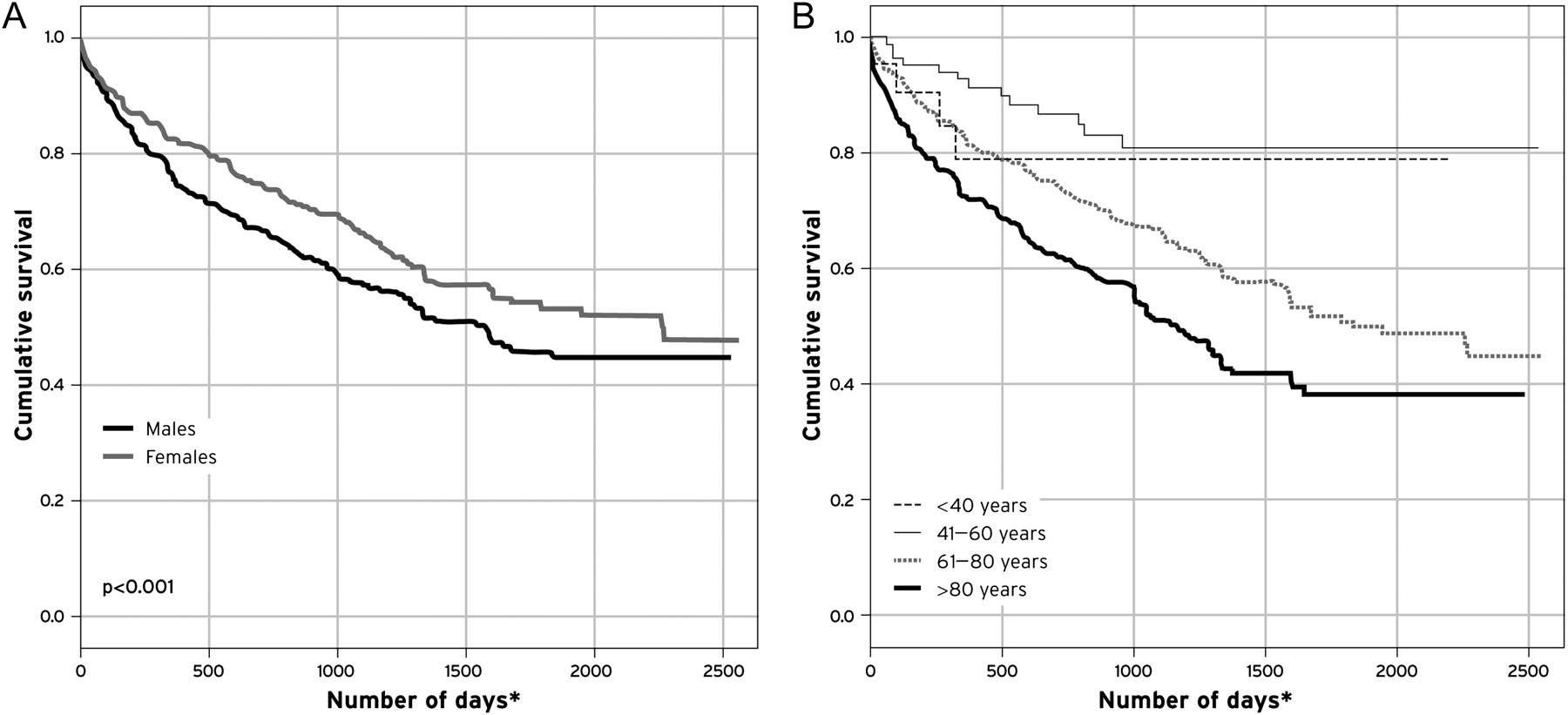
There were independent variables (age, sex and ePASP) associated with an increased risk of mortality in this cohort (p<0.001). The risk of death for patients with moderate pulmonary hypertension (ePASP 50–60 mm Hg) was 1.89 (1.34 to 2.67) compared with mild PHT. An ePASP >60 mm Hg had an increased 3.29-fold risk of death compared with patients with an ePASP between 40 and 50 mm Hg (p<0.001). These variables remained consistent with Cox regression analysis (p=0.002) (see figure 2B). There was a significant relationship between mortality and increasing ePASP (p<0.001) (figure 3B), with an approximately 50% higher mortality in those with PASP >60 mm Hg compared with those with mild PASP. Even those with ‘mild’ pulmonary hypertension had a high mortality of 23% during the follow-up. In an unadjusted model, men fared significantly worse than women (figure 4A, p<0.001). Figure 4B demonstrates the survival differences for each quartile of age, with those over age 60 having significantly increased (p<0.0001) mortality compared with their younger counterparts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Survival for all forms of pulmonary hypertension. (A) Gender; (B) quartiles of age. p<0.001. <40 years; 1780±190 days, 41–60 years; 2148±100 days, 61–80 years; 1631±59 days and >80 years; 1325±66 days. *p<0.0001 for comparison between age quartiles.
Discussion
Using a community-based observational cohort study, we are the first to show the lowest indicative prevalences of all forms of PHT confirmed by a central echocardiography laboratory in a discrete population, with 326 detected cases/100 000 inhabitants of Armadale, Western Australia. PHT due to left heart disease was common and PAH while uncommon, was more frequently detected than previously reported. PHT-related prognosis is poor overall. However, in our study PAH had the best prognosis, probably owing to disease-specific treatment. Increasing PASP and age were associated with a higher risk of mortality.
The true prevalence of PAH is uncertain and previous estimates vary from 5 to 52 cases/million population.7–11 Data from the REVEAL registry reported low estimates of PAH prevalence (group 1) at 12.4/million. REVEAL confirmed the changing age demographic within populations of patients with PAH.7 A Scottish registry examined the prevalence of PAH from two perspectives: all admissions to hospital with PAH (1986 and 2001) for adults aged 16–65 years and patients from the Scottish pulmonary vascular unit (1997–2006).9 The hospitalisation data provided a higher prevalence of PAH (52 cases/million population) than the data from expert centres (15–26 cases/million population).8 ,9 In the French registry, the prevalence of PAH was 15 cases/million population.9 Our indicative prevalence of 151 cases per million is higher than the number in these reports. For iPAH, the prevalence reported from the French registry is 6.5 per million, from the Scottish data 25 per million9 and from our data 48 per million. In 1991, the National Institutes of Health in the USA commissioned a national registry of patients with PAH.11 The incidence of ‘primary’ PAH was estimated at 1–2 cases/million population. These data are now 20 years old and it is likely that the figures quoted represent a significant underestimate of the true incidence of iPAH.
Our figures are higher than those of previous reports and possibly reflect the (1) community-based nature of this study; (2) specificity associated with close, retrospective interrogation of patient databases; (3) possible earlier capture of disease before hospital presentation and (4) unrestricted age allowed in our study compared with the Scottish Registry. Previous reports have assessed prevalence using patient referrals to specialist centres8 and thus probably underestimate the true prevalence of PAH within the community. Awareness and recognition of the earlier phases of PAH is low among doctors, with delays in diagnosis reported to be between 2.8 and 3.8 years.10 ,21 The wide range in the reported prevalence of PAH is probably due to differences in the methods, the criteria used to make a diagnosis and to differences in the selected patient populations.
In our study, we have shown the high detection of cases of more common forms of PHT (groups 2–5). Comparative epidemiology data on all forms of PHT are not available. Our study is therefore the first to document estimated cumulative indicative prevalence for all PHT subtypes from a community-based echocardiography population. PHT is present in up to 70% of those with severe left ventricular systolic/diastolic dysfunction,2 and PAP incrementally predicts death in patients with heart failure.22 In patient populations with respiratory conditions such as chronic obstructive pulmonary disease, PHT may affect up to 50%.2 The prevalence of chronic thromboembolic pulmonary hypertension is unclear, with reports ranging from 0.5% to 3.8% in those with acute pulmonary embolism and can occur without previous embolic clinical events.2 Clearly, PHT in all its forms is a clinical feature of many diseases. We therefore highlight the need for increased vigilance and research in all PHT subtypes and beyond a strong focus on PAH.
We report median mortality (from first pressure estimation) of just over 4 years with varying (borderline significance) survival among different PHT subtypes. Median time to death was similar between patients with PHT due to respiratory disease, left heart disease or unknown cause (all around 4 years). Interestingly, those with PAH (group 1) who had been exposed to modern PAH-specific treatment had the highest median survival times (around 5 years), an improvement compared with historical NIH data.11
A large community-based population study recently described small age-related increases in PASP and the marked relationship between increasing PASP and mortality (unadjusted HR=4.65 per 10 mm Hg; p<0.001). This is striking given nearly all of the patients, aged 45–96 years, had a ‘normal’ PASP of <40 mm Hg at any time during that study.23 This was a referred cohort and not a random sample of the population. However, consistent with the Olmstead County data, we found increasing mortality with higher PASP, regardless of cause.
Our study identifies a large group of patients with PHT with no obvious underlying cause from available data. While clearly a cause must exist, this substantive group of patients need further investigation. These data are similar to those from an Italian report of echocardiography-evaluated patients who had an increase in TR velocity > 3m/s, of whom 10.05% were identified with no known cause of the PHT compared with our 15%.20 The indicative PHT rate of 326 cases per 100 000 from our study is likely to be the minimum population prevalence, since patients with PHT of unknown cause were not included in the calculations. In addition, patients with insufficient TR to estimate RVSP might have had PHT, but were not included in the analysis. We cannot determine from our data the number of individuals with PHT but insufficient TR to measure RVSP. Some individuals with PHT in the community might not yet have had echocardiography, or had echocardiography performed outside our catchment area and thus were not included in our analysis.
These data emphasise that PHT (or an elevated ePASP) is not a diagnosis in itself and requires thorough assessment to determine its cause. Doppler echocardiography is a reliable means of assessing PASP non-invasively and, correlates well with RHC24–28 and is the most widely used screening tool for PAH.2 ,29 Only after investigation for other causes and use of RHC, can PAH be diagnosed. This is particularly important since treatment may alter the long-term outcomes for these patients.30–36
This study has a number of limitations. This is a retrospective cohort surveillance study and data on the indicative prevalence of PHT are based on the records of one large specialist centre obtaining the echocardiograms included in this study. More definitive data on the population prevalence would require prospective screening of a large number of individuals from the community being studied. Echocardiography is an imperfect tool for estimating the true PASP, with a recent meta-analysis37 correlating ePASP only modestly with RHC. Correlation with RHC-derived PASP may be improved by only accepting well-aligned TR velocities with a complete Doppler envelope, as was our practice in this study. Further, we have shown a strong association between the degree of echo-derived PASP estimation and mortality and good agreement between our echo data and those patients receiving RHC in our study.
There was a large group (15%) of patients in whom the cause of PHT was not known. Further, where there was insufficient tricuspid regurgitation to estimate the PAP, it is possible PHT might be present, but undiagnosed. Therefore, there may be a selection bias towards under-reporting of the true population prevalence of PHT in this study. Indeed, assuming the same background prevalence of PHT in this group as we found in the group with measurable TRV, the population prevalence of PHT may be closer to 430/100 000 individuals. Conversely, the lack of tricuspid regurgitation may reflect a lower prevalence of PHT in this population and improved methods of detection of PHT, without reliance on tricuspid regurgitation, are needed in the future. In order to calculate the ‘indicative’ prevalence of PHT we used a conservative approach by adopting a denominator that included a much wider catchment area than Armadale alone, reflecting the regions serviced by our echocardiography laboratory. We acknowledge that using a cut-off point of PASP ≥40 mm Hg has the potential to miss mild to moderate PHT. However, previous work by Mukerjee et al showed that 97% of patients with PAH have a Doppler echocardiographic PASP ≥45 mm Hg, and a threshold of <40 mm Hg versus ≥40 mm Hg has a positive predictive value of 92% and negative predictive values of 44%.28
In summary, this is the first study to assess the ‘indicative’ prevalence of all forms of PHT in a community-based echocardiology laboratory. Using a retrospective database review of all patients undergoing Doppler echocardiographic measures with an elevated PASP (≥40 mm Hg), we found that PHT is a common and dangerous disease. The estimated minimum prevalence of all forms of PHT in this community was 326 cases/100 000 population and for PAH specifically (group 1) the minimum prevalence was 15/100 000 population. We found the estimated median survival in this population to be about 4 years. Identification of the underlying cause may trigger disease-specific treatment, with significant prognostic implications. Confirmation of our data requires further prospective population-based research in addition to more detailed prospective national or international all-cause PHT registries.
References
Footnotes
Funding DP received an unrestricted research grant from Actelion Pharmaceuticals. No company was involved in the collection, analysis or reporting of these data. All authors contributed to the development, design and review of this manuscript.
Competing interests GS receives honoraria from the Pulmonary Hypertension Society of Australia and New Zealand (PHSANZ). The PHSANZ receives grant funding from Actelion, Bayer, GSK, Lilly, Novartis and Pfizer Australia. DP has received honoraria for speaking from Actelion and Pfizer Australia. SS, JD, HN, AK have no conflict of interest to declare. EG has received research funds from Actelion, Bayer, GSK and Pfizer Australia. EG has received honoraria for speaking from Actelion, Bayer, GSK and Pfizer. EG in currently a member of the Advisory Boards for Actelion, Eli Lilly, GSK and Pfizer Australia.
Ethics approval Ethics approval was provided by University of Notre Dame ethics comittee.
Provenance and peer review Not commissioned; externally peer reviewed.