Article Text

Abstract
Objectives Compare the effect of apixaban and warfarin on coagulation and primary haemostasis biomarkers in atrial fibrillation (AF).
Methods The biomarker substudy from the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation trial included 4850 patients with AF randomised to treatment with apixaban or warfarin. Sixty per cent of patients used vitamin K antagonist (VKA) within 7 days before randomisation. Prothrombin fragment 1+2 (F1+2), D-dimer, soluble CD40 ligand (sCD40L) and von Willebrand factor (vWF) antigen were analysed at randomisation and after 2 months of study treatment.
Results In patients not on VKA treatment at randomisation, F1+2 and D-dimer levels were decreased by 25% and 23%, respectively, with apixaban, and by 59% and 38%, respectively, with warfarin (p<0.0001 for treatment differences for both). In patients on VKA at randomisation, F1+2 and D-dimer levels increased by 41% and 10%, respectively, with apixaban and decreased by 37% and 11%, respectively, with warfarin (p<0.0001 for treatment differences for both). sCD40L levels were slightly increased at 2 months, regardless of VKA or randomised treatment. Apixaban and warfarin also both reduced vWF antigen regardless of VKA treatment. The efficacy (stroke) and safety (bleeding) of apixaban compared with warfarin was similar irrespectively of biomarker levels at 2 months.
Conclusions Treatment with apixaban compared with warfarin for stroke prevention in patients with AF was associated with less reduction in thrombin generation and fibrin turnover. This effect of apixaban could contribute to the clinical results where apixaban was superior to warfarin both in stroke prevention and in reducing bleeding risk.
Trial registration number NCT00412984.
- atrial fibrillation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Atrial fibrillation (AF) is associated with increased risk of thromboembolic events. The reduced flow in the left atrial appendage and in the atrium, in combination with platelet activation and endothelium dysfunction, increase coagulation activity and contribute to the risk of stroke.1 2 The concentrations of prothrombin fragment 1+2 (F1+2), and D-dimer, markers for thrombin generation and fibrin turnover, are increased in AF and reduced by warfarin treatment.3 Higher D-dimer levels are also associated with increased risk of future cardiovascular events.4 5 CD40 ligand is stored in the alpha-granules and expressed at the time of platelet activation. Soluble CD40 ligand (sCD40L), a part of the CD40L receptor, which is released and shed from activated platelets, exhibits elevated concentrations in AF.6 Endothelial injury also contributes to the activation of primary haemostasis in AF, which might be reflected by increased levels of von Willebrand factor (vWF).7 Both sCD40L and vWF have been described to be associated with future cardiovascular events in patients with AF without anticoagulant treatment.8 9
In the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial, the direct factor Xa (FXa) inhibitor, apixaban, was superior to warfarin in prevention of stroke/systemic embolism (SE) and simultaneously reduced the risk of bleeding in patients with AF.10 In acute deep venous thrombosis and acute coronary syndromes, where there is a high level of coagulation activity due to ongoing thrombus formation, apixaban has been shown to reduce indicators of coagulation activity.11 12 In experimental models, apixaban also reduced tissue factor-induced platelet aggregation in a dose-dependent manner.13 There is, however, limited knowledge of the effects of apixaban on coagulation and platelet biomarkers in AF when the treatment is given as prevention rather than treatment of a thrombotic event. The aims of this study were to evaluate the effect of apixaban compared with warfarin on biomarkers reflecting coagulation activity, platelet activation and endothelial injury, and to evaluate the efficacy and safety of apixaban compared with warfarin in relation to the levels of these biomarkers during treatment.
Methods
The ARISTOTLE trial
The details of the ARISTOTLE trial have been published previously.10 Briefly, ARISTOTLE was a double-blind, double-dummy, randomised clinical trial that enrolled patients with AF and at least one CHADS2 risk factor for stroke/SE. Patients were randomly assigned (1:1) to receive either warfarin (target international normalised ratio (INR) 2.0–3.0) or apixaban (5 mg twice daily). The trial included 18 201 patients and the median follow-up time was 1.8 years. The primary efficacy end point was stroke/SE and the primary safety outcome was major bleeding according to the International Society of Thrombosis and Haemostasis classification.14
Biochemical analyses
All patients participating in the ARISTOTLE trial were invited to take part in blood sampling for the biomarkers substudy at baseline. At selected sites, around 5000 patients were preplanned for invitation to participate in the serial blood sampling substudy with repeated sampling at the 2-month visit. This population constituted the material of the current study. Among the 4850 patients included in the serial biomarkers substudy, 4847 had measurements for vWF antigen, 4849 for F1+2 and sCD40L and all 4850 for D-dimer.
Blood samples for core laboratory analyses of prespecified serial biomarkers were obtained at randomisation before start of study treatment and after 2 months of randomised treatment. After centrifugation at 2000×g for 15 min, plasma was stored at −80°C until analysis. Enzyme immunoassays were used for the analyses of F1+2 (Enzygnost F1+2, Siemens, Germany), D-dimer (Asserachrome, Stago, France), sCD40L (Human sCD40L, Bender MedSystems, GmbH) and vWF antigen (STA-Liatest vWF, Stago, France) at the Uppsala Clinical Research Center laboratory, Sweden. The reference intervals were 69–229 pmol/L for F1+2, <500 µg/L for D-dimer, 0.06–3.98 µg/L for sCD40L and 0.60–1.60 kIE/L for vWF antigen. The interassay precisions were 9%, 11%, 9% and 4% for F1+2, D-dimer, sCD40L and vWF antigen, respectively.
Clinical end points and risk factor classification
The end points for this study included stroke/SE, death of all causes, major bleeding and clinically relevant non-major (CRNM) bleeding.15 All clinical events were classified by a blinded clinical event committee using prespecified criteria.15 CHA2DS2-VASc and HAS-BLED scores were calculated individually for each patient based on the sum of risk factors present at randomisation. Patients were categorised by CHA2DS2-VASc according to score 0–1, 2, 3, 4 and ≥5, and by HAS-BLED according to score 0–1, 2 and ≥3.
Statistical analyses
Baseline characteristics were reported by treatment arm using median and 25th and 75th percentiles for continuous variables and frequencies and percentages for categorical variables. Comparisons of biomarker levels at randomisation and 2 months between the apixaban and warfarin groups were stratified by randomised treatment and use of vitamin K antagonist (VKA) treatment within 7 days before randomisation, using box plots showing medians and 25th and 75th percentiles with whiskers showing 10th and 90th percentiles. Patients not treated with VKA within 7 days before randomisation to study drug were defined as the no VKA group. Patients receiving treatment with VKA within 7 days before randomisation to study drug make up the VKA group. Randomised treatment groups were compared by use of VKA treatment at baseline using a linear regression model with the logarithm of the 2-month biomarker levels as dependent variables and logarithm of the baseline biomarker levels and randomised treatment as independent variables. The biomarker levels were log-transformed before analysis due to skewed distribution. Consequently, model-adjusted estimates were presented as geometric means and treatment group differences as ratios of geometric means.
Primary and secondary efficacy end point analyses of time from the 2-month measurement to clinical outcomes or censoring included all randomised substudy patients and all first events occurring between the 2-month measurement and the efficacy cut-off date (31 January 2011). Analyses of bleeding outcomes were based on the on-treatment population including all randomised patients enrolled in the substudy who received at least one dose of the study drug and included all first events occurring between the 2-month measurement and 2 days after the last dose of the study drug. For patients experiencing events within 2 months of randomisation, the first event occurring after 2 months was counted as the first event.
The relations between clinical outcomes after 2 months, study treatment and biomarker levels at 2 months were analysed using a Cox regression model including treatment group, biomarker and the treatment by biomarker interaction as covariates. Biomarker levels were analysed as categorical variables (under/above median) and as continuous log transformed variables, fitted using restricted cubic splines. The median levels of biomarkers at 2 months were 75.9 pmol/L, 401 µg/L, 0.62 µg/L and 1.23 kIE/L for F1+2, D-dimer, CD40L and vWF antigen, respectively. Event rates per 100 patient-years of follow-up were reported. The total patient-years of follow-up was 8907.
All analyses were performed using SAS software, V.9.4 (SAS Institute, Cary, North Carolina, USA). Results with a two-sided p value <0.05 were considered to be statistically significant, and as all analyses were considered exploratory there were no adjustments for multiple comparisons.
Results
Baseline characteristics
Baseline characteristics in relation to treatment are presented in table 1. Mean age of patients was 69.0 (SD 9.4) years, 34.1% (n=1654) were women and a majority of patients (83.8%, n=4062) had permanent or persistent AF. The distributions of risk factors, risk scores and medical treatment were similar to the main ARISTOTLE population.10 The no VKA and VKA groups included 1946 and 2894 patients, respectively. In the VKA group, the mean time in therapeutic range of the international normalized ratio (INR) during the study was 64%.
Baseline characteristics of the biomarkers substudy cohort
Thrombin generation and fibrin turnover during apixaban or warfarin treatment
The baseline levels of F1+2 were higher in the no VKA group compared with the VKA group (figure 1A). Patients in the no VKA group treated with apixaban for 2 months (no VKA/apixaban group) had a mean reduction of 25% in F1+2 levels compared with baseline, with a ratio of month 2/baseline of 0.75 (95% CI 0.70 to 0.80) (figure 1A; table 2). In contrast, patients in the VKA group treated with apixaban for 2 months (VKA/apixaban group) had a 41% mean increase in F1+2 levels compared with baseline, estimated ratio month 2/baseline 1.41 (95% CI 1.34 to 1.48) (figure 1A; table 2). Treatment with warfarin for 2 months, reduced F1+2 levels in the no VKA and VKA groups by 59%, ratio month 2/baseline 0.41 (95% CI 0.38 to 0.43) and 37%, ratio month 2/baseline 0.63 (95% CI 0.60 to 0.66), respectively (figure 1A; table 2). Accordingly, treatment with warfarin was associated with significantly larger reductions and lower F1+2 levels than apixaban regardless of VKA use at randomisation (p<0.0001). Ratios of geometric means of F1+2 levels after apixaban versus warfarin treatment were 1.84 (95% CI 1.67 to 2.02) and 2.24 (95% CI 2.09 to 2.41) for the no VKA and VKA groups, respectively.
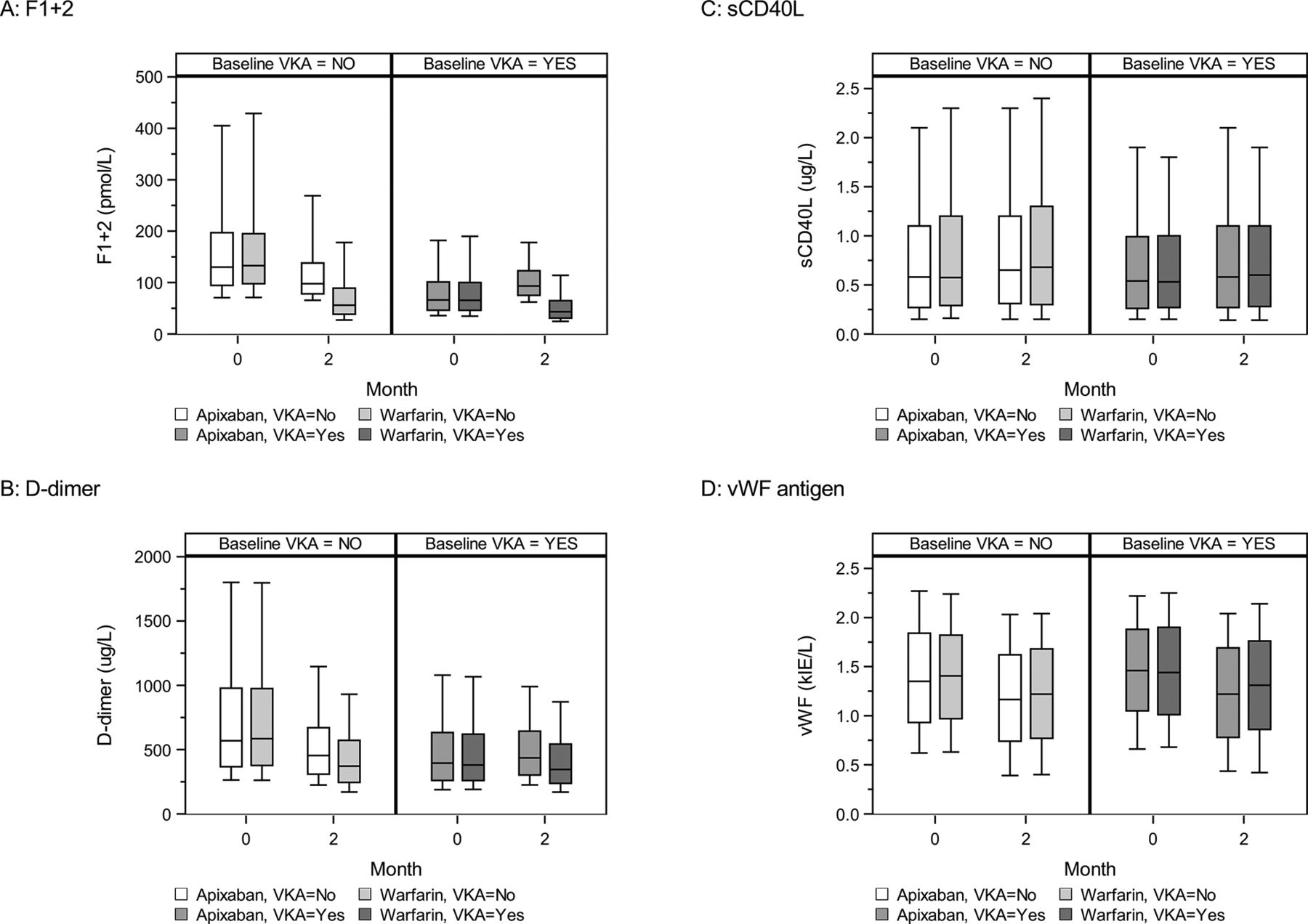
The concentrations of fragment 1+2 (F1+2) (A), D-dimer (B), soluble CD40 ligand (sCD40L) (C) and von Willebrand factor (vWF) antigen (D) in the apixabanand warfarin treatment groups stratified by warfarin (vitamin K antagonist (VKA)) status at randomisation. Boxes represent the 25th–75th percentiles with the median marked by a black line. Whiskers represent the 10th and 90th percentiles.
Biomarker concentrations at month 2, by baseline vitamin K antagonist treatment group
The baseline level of D-dimer was higher in the no VKA group compared with the VKA group (figure 1B). Apixaban treatment for 2 months was associated with a 23% mean reduction in D-dimer levels in the no VKA group compared with baseline, estimated ratio 0.77 (95% CI 0.74 to 0.80) (figure 1B; table 2). In contrast, the VKA/apixaban group had a 10% mean increase in D-dimer level, estimated ratio 1.10 (95% CI 1.07 to 1.12). Warfarin was associated with a 38% mean reduction of D-dimer levels in the no VKA group, estimated ratio 2 months/baseline 0.62 (95% CI 0.60 to 0.64) while the VKA/warfarin group exhibited an 11% mean reduction, ratio 2 months/baseline 0.89 (95% CI 0.86 to 0.91) (figure 1B; table 2). Accordingly, treatment with warfarin was associated with significantly larger reductions and lower D-dimer levels than apixaban regardless of VKA use at randomisation (p<0.0001). Ratios of geometric means of D-dimer levels after apixaban versus warfarin treatment were 1.24 (95% CI 1.18 to 1.31) and 1.24 (95% CI 1.20 to 1.28) for the no VKA and VKA groups, respectively.
Biomarkers for primary haemostasis during apixaban or warfarin treatment
The concentrations of sCD40L and vWF antigen were similar in the no VKA and VKA groups at baseline (figure 1C,D). Levels of sCD40L increased by 9% in the no VKA/apixaban group compared with baseline, with a ratio month 2/baseline of 1.09 (95% CI 1.04 to 1.14), whereas sCD40L levels increased by 5% in the VKA/apixaban group (figure 1C; table 2). Similar results were found in the group randomised to warfarin treatment (figure 1C; table 2). There were no significant differences of sCD40L concentrations comparing the apixaban and warfarin treatment groups at 2 months (p=0.5), regardless of VKA treatment.
Apixaban treatment for 2 months reduced vWF antigen by 20% in the no VKA group (estimated ratio 0.80 (95% CI 0.77 to 0.84)) and by 18% in the VKA group (estimated ratio 0.82 (95% CI 0.79 to 0.84)), respectively (figure 1D; table 2). Similar results were found in the no VKA and on VKA groups when randomised to warfarin treatment. No significant differences in vWF antigen levels at 2 months was found between apixaban/warfarin groups regardless of VKA treatment.
Clinical outcomes in relation to biomarker concentrations during treatment with apixaban or warfarin
From 2 months after study start until the end of follow-up, 43 patients in the apixaban group (0.96 %/year) experienced a stroke/SE compared with 53 patients (1.23 %/year) in the warfarin group. Major/CRNM bleeding occurred in 169 (4.05%/year) and 210 (5.34%/year) patients in the apixaban and warfarin groups, respectively. The efficacy and safety of apixaban compared with warfarin as assessed by the clinical end points was not significantly different across any of the biomarker concentrations at 2 months (figure 2).

{kind=link}
{kind=link}
Efficacy of apixaban relative to warfarin for fragment 1+2 (F1+2) and D-dimer levels at 2 months (A). Efficacy of apixaban relative warfarin for soluble CD40 ligand (sCD40L) and von Willebrand factor (vWF) levels at 2 months (B). Cox proportional hazards model with biomarker level, treatment and interaction between biomarker level and treatment as covariates. CRNM, clinically relevant non-major; SE, systemic embolism.
Discussion
The present study showed that in patients with AF, apixaban treatment reduced coagulation activity, as indicated by reduction in plasma concentrations of biomarkers reflecting thrombin generation and fibrin turnover. The reduction in coagulation activity with apixaban was less than with warfarin. Both anticoagulant treatments were associated with only minor changes in the levels of the platelet activity indicators sCD40L and vWF antigen. In the main ARISTOTLE trial, apixaban was superior to warfarin in prevention of stroke/SE, and simultaneously reduced the risk of bleeding in patients with AF. The less pronounced decrease of the coagulation activity during apixaban treatment compared with warfarin treatment may contribute to the clinical results.
Apixaban treatment reduced F1+2 levels in this cohort of patients with AF, as has previously been described in patients with ongoing acute thromboembolism.11 With its selective inhibition of coagulation FXa, the reduction in F1+2 levels was moderate compared with warfarin. The F1+2 levels measured in this study were in the upper range of the reference interval at baseline and the optimal cut-off level for F1+2 for identification of patients with AF at low risk of stroke remains to be defined. Apixaban reduced F1+2 levels to around the median concentration of the reference interval. Similar results of a moderate reduction of F1+2 have recently been described for rivaroxaban in patients with cardioembolic stroke, where prophylactic treatment with dabigatran did not affect F1+2 levels.16 17 Low F1+2 concentrations, however, as seen during warfarin therapy, might be associated with a decreased ability to protect against bleeding events and could therefore increase the risk of bleeding without substantial association with lower risk of stroke/SE.18 Elevated F1+2 levels have been described in stroke patients with AF, and as F1+2 levels reflect thrombin generation they have been suggested as a better marker for stroke prediction than biomarkers including fibrin turnover.19 The levels of F1+2 during oral anticoagulant (OAC) treatment in the present study were within the normal reference interval, indicating that a moderate decrease by apixaban could be sufficient to protect against stroke without limiting the sufficient residual thrombin available for clot formation as a protective mechanism against bleeding. This hypothesis however needs further evaluation.
D-dimer was also effectively reduced after 2 months of apixaban treatment. Only a small proportion of patients had D-dimer concentrations above the reference interval during OAC treatment. The effect of apixaban on D-dimer concentrations in this AF cohort, without an acute thromboembolic event, was similar to results described in studies of venous thromboembolism.11 Treatment with rivaroxaban has been found to reduce D-dimer levels similar to the results found for apixaban in the present study.20 In contrast, 150 mg of dabigatran was found to have a more pronounced effect on D-dimer compared with warfarin in the Randomized Evaluation of Long-Term Anticoagulation Theraphy (RELY) trial.21 High D-dimer levels before start of OAC treatment have been used for successful identification of patients at increased risk of stroke/SE and bleeding.4 D-dimer is a marker for thrombin generation and a marker for endogenous fibrin turnover, and levels are influenced by age, sex, underlying vascular disease and inflammation; factors associated with patient frailty.22 Why high D-dimer is associated with increased risk of bleeding is not fully explored, but fibrinogen formation is important when bleeding occurs and an enhanced fibrin turnover, resulting in increased D-dimer levels, might thus be disadvantageous.23
Patients that had been exposed to warfarin/VKA before randomisation and then received warfarin as study drug exhibited further reduced levels of F1+2 and D-dimer. Patients receiving warfarin were regularly monitored during the trial, which could have improved the time in therapeutic range of -INR, which is associated with D-dimer levels and with risk of stroke in patients with AF.3 24
Although thrombus formation mechanisms in AF are not fully elucidated, the endothelium of the left atrial appendage is activated and responds to stimuli to a higher degree than in the right appendage.1 The rhythm and the low flow in the appendage increase platelet activity and formation of microparticles, which also contribute to an increased coagulation potential in AF.2 19 In this study, the platelet activity marker sCD40L was not reduced after 2 months of apixaban or warfarin treatment. In contrast, concentrations were slightly increased compared with baseline in both treatment groups, regardless of VKA treatment at randomisation. The concentration of sCD40L was in the reference interval in the majority of the patients and there might be a need for a more potent activation of platelets, including thrombin formation, to observe an effect on sCD40L by OAC treatment. Limited data exist regarding the effect of direct FXa inhibitors on platelet activity and how the activity should be evaluated. Edoxaban inhibits platelet aggregation in vitro to a higher degree compared with fondaparinux.25 To further evaluate the effect of apixaban on platelet activity in AF, investigations of other platelet markers are needed.26 In contrast to sCD40L, vWF antigen levels were elevated in a proportion of patients independent of VKA status before randomisation. These findings are in accordance with previous results from a small study where warfarin for 6 weeks did not change vWF antigen levels.27 However, apixaban treatment for 2 months reduced the vWF antigen by 20% and similar results were found for warfarin study treatment. vWF antigen is associated with increased risk of stroke and venous thrombosis and whether FXa inhibition could modulate the dysregulation of vWF antigen found in thromboembolic diseases has to be determined.
The risk of intracerebral bleeding is lower with all non-vitamin K oral anticoagulants compared with warfarin.10 28–30 The mechanism behind this might be that the available amount of FVIIa is enough to form thrombin on injury. The rapid initiation of coagulation by the tissue factor/FVIIa complex forming thrombin is indeed of high importance in organs such as the brain. In the RELY trial, the direct thrombin inhibitor dabigatran induced only a minor reduction of FVIIa concentrations compared with warfarin.21 Similar results with only minor reduction of FVIIa can also be expected by treatment with inhibitors that are selectively targeting FXa. FVIIa concentrations might thus constitute a valuable future biomarker for predicting intracerebral bleeding during OAC treatment.
Limitations of the present study include that the values for time in therapeutic range of INR before randomisation were not known and that the prespecified end point stroke included both ischaemic and haemorrhagic stroke. Due to few events we did not categorise the end point further. As the ARISTOTLE trial contained exclusion criteria such as severe renal dysfunction, not all patients with AF were eligible for participation. The study population may therefore not be representative for unselected patients with AF. This might have influenced the concentrations of the coagulation activity markers in the present study and the concentrations might be different in patients with AF in everyday clinical practice.
In conclusion, apixaban reduced the coagulation activity as measured by F1+2 and D-dimer, to a lesser degree than warfarin. Apixaban and warfarin treatment both reduced vWF antigen as a measurement of endothelial injury but had no effect on sCD40L. The effect of apixaban compared with warfarin for the risk of future thromboembolic and bleeding events was consistent at all biomarker levels.
Key messages
What is already known on this subject?
Atrial fibrillation is associated with a prothrombotic state including the endothelium, platelets and plasma coagulation.
In acute situations such as deep venous thrombosis or acute coronary syndrome with an increased coagulation activity, apixaban has been shown to reduce indicators for plasma coagulation.
What might this study add?
In a situation without an acute increased coagulation activity such as in atrial fibrillation, apixaban reduces coagulation markers D-dimer and fragment 1+2 (F1+2) to a lower degree compared with warfarin, which seems enough to protect against stroke without increasing risk for bleeding.
Apixaban and warfarin treatment in atrial fibrillation did not change CD40L and von Willebrand factor as markers for platelet and endothelial activation.
How might this impact on clinical practice?
Further broaden the understanding of how apixaban compared with warfarin influences coagulation activity in atrial fibrillation and that the more moderate reduction of D-dimer and F1+2 may be preferable to balance the risk-benefit of oral anticoagulant treatment.
Acknowledgments
The authors would like to thank Ebba Bergman, PhD, and Vendela Roos, PhD, at Uppsala Clinical Research Center (UCR), Uppsala University, Uppsala, Sweden for providing editorial assistance.
References
Footnotes
Contributors CC, LW and AS contributed substantially to the design of the present biomarker substudy in the ARISTOTLE trial. CC, LW, AS, JHA, CBG, MH, BJG, RDL and EMH contributed to the design of the ARISTOTLE biomarker substudy as members of the ARISTOLE biomarker substudy committee. CC, KH, RDC, MA, B-HO and SH contributed to the acquisition of data as ARISTOTLE investigators. AS was responsible for the plasma measurements of F1+2, D-dimer, sCD40 ligand, and vWF at the UCR laboratory. UA performed the statistical analyses. CC and AS provided the first draft of the manuscript. All coauthors participated in the interpretation of data and critically revised the manuscript. All authors have approved the final version of the manuscript. The first (CC) and senior (AS) authors and the statistician (JHA) had full access to the database and take responsibility for the integrity and of the data and the data analyses.
Funding The ARISTOTLE trial was funded by Bristol-Myers Squibb, Princeton, New Jersey and Pfizer, New York, New York, and coordinated by the Duke Clinical Research Institute (DCRI), USA, and by Uppsala Clinical Research Center (UCR), Uppsala, Sweden.
Competing interests CC: lecture fees from Boehringer Ingelheim, Bristol-Myers Squibb, CSL Behring, Novartis. LW: institutional research grants from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/Pfizer, GlaxoSmithKline, Merck & Co, Roche Diagnostics; holds two patents involving GDF-15. UA: institutional research grant from Bristol-Myers Squibb/Pfizer. JHA: research grants from Boehringer Ingelheim, Bristol-Myers Squibb, CSL Behring, FDA, NIH, CryoLife, Tenax Therapeutics, VoluMetrix; honoraria from Bristol-Myers Squibb, CSL Behring, Janssen Pharmaceuticals, Merck, NovoNordisk Pharmaceuticals, Pfizer, VA Cooperative Studies Program, Zafgen; consultancy/advisory board fees from AbbVie, Bristol-Myers Squibb, CSL Behring, Janssen Pharmaceuticals, Merck, NovoNordisk Pharmaceuticals, Pfizer, Portola Pharmaceuticals, VA Cooperative Studies Program, Zafgen. MA: speaker’s bureau from Pfizer; honoraria from Pfizer; consultant/advisory board from Bristol-Myers Squibb, Daiichi Sankyo, Bayer, Boehringer Ingelheim, Pfizer. RDC: research grants from Bayer, Bristol-Myers Squibb/Pfizer, Daiichi Sankyo; speaker’s bureau from Daiichi Sankyo, Bristol-Myers Squibb/Pfizer; honoraria from Daiichi Sankyo, Bayer, Bristol-Myers Squibb/Pfizer; consultant/advisory board from Bayer, Daiichi Sankyo. BJG: data safety monitoring board from Boston Scientific, Cardiovascular Research Foundation, Duke Clinical Research Institute, Duke University, Icahn School of Medicine at Mount Sinai, Janssen Research & Development, Kowa Research Institute, Medtronic, Mount Sinai St Lukes, Teva Pharmaceutical Industries, Myokardia; consultancy fees from Coretherapix SLU, Janssen Scientific Affairs, Sirtex Medical. CBG: research grants from Armetheon, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo; Duke Clinical Research Institute, FDA, GlaxoSmithKline, Janssen Pharmaceuticals, Medtronic Foundation, Novartis, Pfizer; consultancy/advisory board fees from AbbVie, Armetheon, AstraZeneca, Bayer, Boehringer Ingelheim, Boston Scientific, Bristol-Myers Squibb, Daiichi Sankyo, Gilead Sciences, GlaxoSmithKline, Janssen Pharmaceuticals, Medscape, Medtronic, Merck, NIH, Novartis, Pfizer, Rho Pharmaceuticals, Sirtex, Verseon. SH: honoraria from AstraZeneca, Bayer, Boehringer Ingelheim, Pfizer, Bristol-Myers Squibb, Merck, Sanofi. MH: former employee of and has stock ownership in Bristol-Myers Squibb. KH: speaker’s bureau from Daiichi Sankyo. EMH: research grants from Janssen Pharmaceuticals; honoraria from Bristol-Myers Squibb/Pfizer, Boehringer Ingelheim; consultancy/advisory board fees from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb/Pfizer, Medtronic, Portola. RDL: research grants from Amgen, Bristol-Myers Squibb/Pfizer, GlaxoSmithKline, Medtronic PLC, Sanofi-Aventis; consultant/advisory board fees from Bristol-Myers Squibb/Pfizer, Bayer, Boehringer Ingelheim. B-HO: grant and personal fees from Novartis. AS: institutional research grants from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/Pfizer, Roche Diagnostics, GlaxoSmithKline; consultancy fees from Olink Proteomics.
Patient consent Not required.
Ethics approval Approval by the appropriate ethics committees was obtained at all sites.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice Since this paper was first published online it has been made open access.