Article Text

Abstract
Objective Sudden cardiac death (SCD) in families with premature atherosclerosis (PAS) is generally attributed to lethal arrhythmias during myocardial infarction. Yet, such arrhythmias may also arise from non-ischaemic inherited susceptibility. We aimed to test the hypothesis that Brugada syndrome is prevalent among families with PAS in which SCD occurred.
Methods We investigated all patients who underwent Ajmaline testing to screen them for Brugada syndrome because of unexplained familial SCD in the Amsterdam University Medical Centers between 2004 and 2017. We divided the cohort into two groups based on a positive family history for PAS. All individuals with a positive Ajmaline test were screened for SCN5A-mutation.
Results In families with SCD and PAS, the prevalence of positive Ajmaline test was similar to families with SCD alone (22% vs 19%). The number of SCD cases in families with SCD and PAS was higher (2.34 vs 1.63, p<0.001) and SCD occurred at older age in families with SCD and PAS (42 years vs 36 years, p<0.001), while the prevalence of SCN5A mutations was lower (3% vs 18%, p<0.05).
Conclusions Brugada syndrome has a similar prevalence in families with SCD and PAS as in families with SCD alone, although SCD in families with SCD and PAS occurs in more family members and at older age, while SCN5A mutations in these families are rare. This suggests that the SCD occurring in families with PAS could be related to an underlying genetic predisposition of arrhythmias, with a different genetic origin. It could be considered to screen families with SCD and PAS for Brugada syndrome.
- atherosclerosis
- brugada syndrome
- ajmaline
- death
- sudden
- cardiac
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Sudden cardiac death (SCD) most often results from lethal cardiac arrhythmia (ventricular fibrillation (VF)) that stems from inherited and acquired causes and combinations of both. When SCD occurs at young age (typically <45 years), the prevalence of acquired causes is still low, increasing the likelihood of an inherited cause. Various inherited cardiac diseases may increase SCD risk because they disrupt electrophysiological or structural properties, thereby increasing the vulnerability to VF. Families in whom SCD at young age occurred are referred for cardiogenetic examination in order to uncover the familial inherited disease and identify relatives who carry that disease and should receive timely treatment to prevent SCD. Conversely, if SCD occurred at older age, relatives of the SCD victim are usually not referred for cardiogenetic investigation, because the likelihood of an inherited disease is considered low. This especially applies to families with atherosclerotic disease, since it is assumed that SCD cases in these families were caused by acute myocardial infarction. This assumption stems from autopsy studies which indicated that coronary artery disease (CAD) is the cause of death in roughly 80% of SCD cases.1 However, these studies are prone to referral bias, since only a minority of SCD cases underwent postmortem investigation. Recently, a large prospective study was conducted, where all SCD cases in the total San Francisco county were investigated. Here it was observed that only 32% of all autopsy confirmed SCD cases were due to CAD.2 These autopsy studies also showed that, among all SCD cases in which CAD was found, only 30%–50% had died of an acute CAD event.2 Moreover, Dutch registries have shown that the risk to die from a myocardial infarction for individuals <65 years of age is less than 10%.3
We hypothesised that inherited cardiac diseases that increase SCD risk also play a role in SCD in families with atherosclerotic disease. In particular, Brugada syndrome (BrS) may be involved, because it shares the same key electrophysiological mechanism that underlies VF with acute myocardial infarction/ischaemia: reduced cardiac excitability. To test this hypothesis, we studied a large consecutive cohort of patients who underwent Ajmaline testing to screen them for BrS because of unexplained SCD in their family and compared the proportion of a positive test and other group properties between families with premature atherosclerosis and families without premature atherosclerosis.
Methods
Data collection
We investigated all patients who underwent Ajmaline tests, conducted as described before,4 at the Amsterdam University Medical Centers (UMC), location AMC, the Netherlands, between December 2004 and December 2017. Information on family history of SCD and premature atherosclerosis were collected from the medical records of the index case. When missing, these individuals were contacted for a questionnaire. Indexes and their family members (defined as all descendants of a common ancestor) were included in the analysis if the Ajmaline test was performed because of unexplained SCD in the family; they were excluded if the Ajmaline test was performed because of a family history of BrS or if the baseline ECG raised the suspicion of BrS in the absence of familial SCD. Since the Amsterdam UMC has the policy that retrospective cohort studies are considered as non-compulsory research, approval of the Medical Ethics Committee was waved for our research protocol. Written informed consent was not obtained, except for patients of whom the pedigree figures are displayed. They all singed a written informed consent that they agreed with publication of their data, as long as they remained anonymous.
Premature atherosclerosis
A family history of premature atherosclerosis was defined as greater than one family member with a myocardial infarction, percutaneous coronary intervention or coronary artery bypass graft in men before 51 years of age and in women before 56 years of age.
Genetic testing
All index patients who had a positive Ajmaline test were offered genetic testing of the SCN5A gene with methods described previously.5
Statistical analysis
Study characteristics and ECG changes after the administration of Ajmaline were analysed and stratified by family history of premature atherosclerosis. We analysed our data both using individual patient data and whole family data separately. A total of 0.2% of the variables were missing and was imputed by multiple imputation using logistic and linear regression models.6 Continuous variables are expressed as mean±SD and categorical variables are expressed as total number with percentage and 95% CI for proportion calculated using the Wilson Method. Student’s t-tests and χ2 test were used to calculate differences in patient and group characteristics with IBM SPSS Statistics for Windows, V.24.0. A p value <0.05 was considered statistically significant.
Results
Inclusion
In total, 1730 individuals underwent Ajmaline testing. A flow diagram of inclusion is shown in figure 1. Of these individuals, 929 were excluded, mostly because they underwent Ajmaline testing because of a family history of BrS or because the ECG raised the suspicion of BrS in the absence of familial SCD. The final study population consisted of 801 individuals from 440 families who underwent Ajmaline testing because of unexplained SCD in the family. Of these 440 families, 43% also had a family history of premature atherosclerosis and 88 (20%) had a positive Ajmaline test. An example of a positive Ajmaline test is displayed in figure 2.

Flow diagram of the inclusion of patients and families with unexplained SCD. BrS, Brugada syndrome; PAS, premature atherosclerosis; SCD, sudden cardiac death.
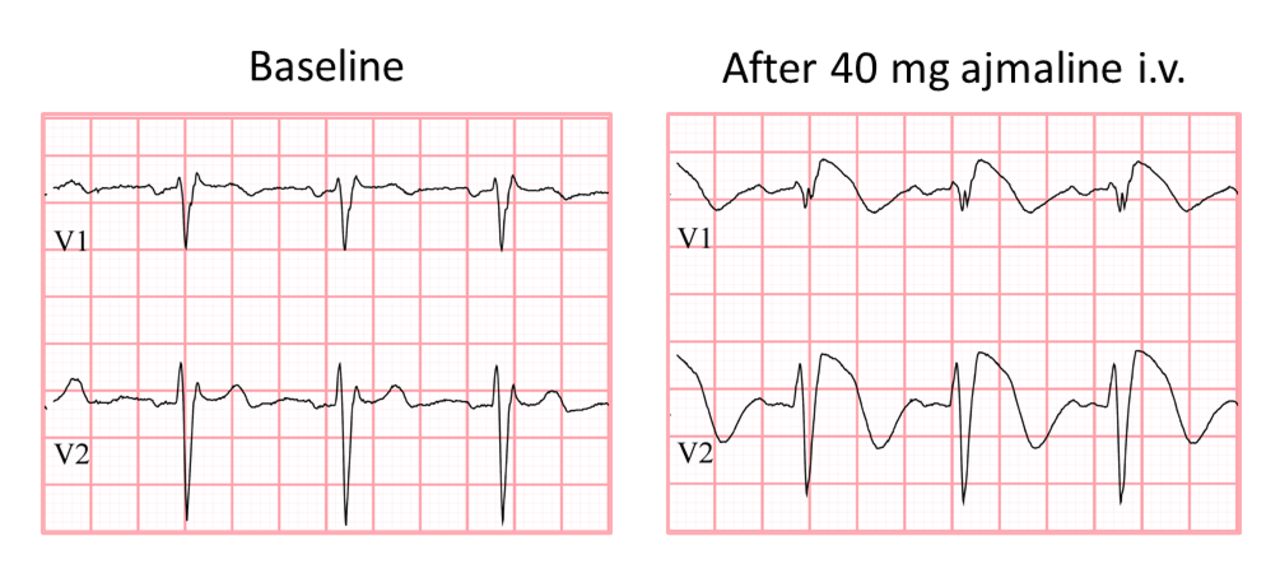
Ajmaline test. Example of positive Ajmaline test in patient with family history of sudden cardiac death.
Proportion of positive Ajmaline tests in families with or without premature atherosclerosis
The proportion of a positive Ajmaline test was similar between families with or without premature atherosclerosis (22% vs 19%, table 1). Families with premature atherosclerosis had more SCD cases and, on average, SCD occurred at older age as compared with families without premature atherosclerosis (42.1 years vs 36.2 years, table 1). The proportion of positive Ajmaline tests was similar in both groups when only families were analysed in which the youngest SCD case occurred at older age; >45 years (16% vs 16%, table 1). We also analysed ECG parameters in individuals after Ajmaline administration since Brugada can be characterised by specific ECG deviations. No significant differences could be found in heart rate, PQ duration or QRS duration between the groups (table 1).
Characteristics of SCD individuals and families with and without premature atherosclerosis
Among the 88 families with a positive Ajmaline test, 80 (91%) indexes agreed to be genetically tested. A pathogenic SCN5A mutation was found in nine families (11%) overall, and in a significantly larger proportion in families without premature atherosclerosis than in families with premature atherosclerosis (8 (18%) vs 1 (3%), respectively; p<0.05).
Properties of families with or without a pathogenic SCN5A mutation
In families with a pathogenic SCN5A mutation, SCD at older age (youngest age within the family >45 years) occurred significantly less often than in families without a pathogenic SCN5A mutation (0% vs 39%), and there were fewer SCD cases within the family (11% vs 68%). Besides, in families with a pathogenic SCN5A mutation, only a small proportion also had a positive family history of premature atherosclerosis (11% vs 49%)(table 2).
Characteristics of all Ajmaline positive tested individuals divided by pathogenic SCN5A mutation.
Pedigrees of families with both SCD and premature atherosclerosis
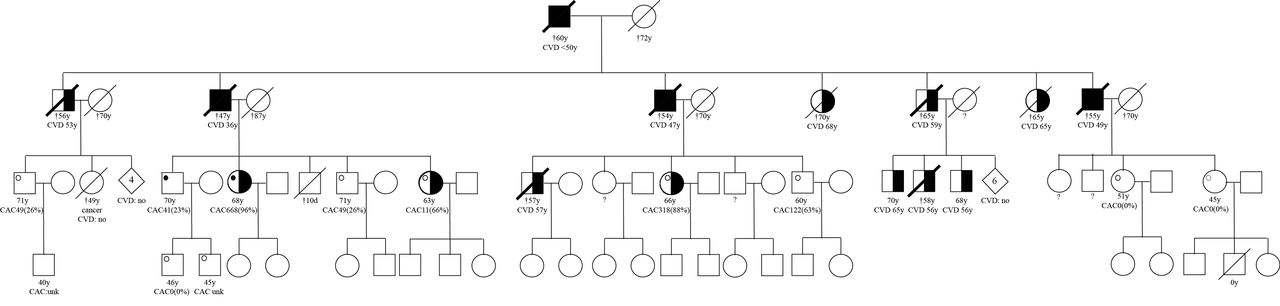
To get an impression about the inheritance patterns, we created pedigrees of the largest families in which more than three individuals underwent Ajmaline testing. Family A, is the largest family, consisting of 29 individuals of which the oldest alive generation that was available for screening. Nine of these individuals underwent Ajmaline testing. Of these two individuals had a positive test and seven had a negative test. No cosegregation was found between a positive Ajmaline test and atherosclerosis (figure 3). In family B, eight individuals of the same generation were available for screening. Out of these eight individuals, six underwent an Ajmaline test. Two individuals had a positive test and one individual with a positive test; also the children were tested, who turned out to be negative. Again no cosegregation could be observed between a positive Ajmaline test and atherosclerosis (figure 4).

Pedigree A squares represent males and circles represent females. Black-filled symbols represent cases with premature atherosclerosis. Half-filled symbols represent cases with atherosclerosis at older age. White symbols represent individuals without atherosclerosis. Diamond symbols represent a summary of individuals (both sexes) and their indicated number. If a symbol is crossed with a thin line this means the individual died. If a symbol is crossed with a thick line, this visualises that individual died suddenly before the age of 65. A black dot represents a positive Ajmaline test, a white dot represents a negative Ajmaline test. CACx(%), coronary artery calcium score with the absolute score and the percentile in parentheses.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pedigree B squares represent males and circles represent females. Black-filled symbols represent cases with premature atherosclerosis. Half-filled symbols represent cases with atherosclerosis at older age. White symbols represent individuals without atherosclerosis. Diamond symbols represent a summary of individuals (both sexes) and their indicated number. If a symbol is crossed with a thin line this means the individual died. If a symbol is crossed with a thick line, this visualises that individual died suddenly before the age of 65. A black dot represents a positive Ajmaline test, a white dot represents a negative Ajmaline test. CACx(%), coronary artery calcium score with the absolute score and the percentile in parentheses.
Discussion
In this study, we observed that in families with premature atherosclerosis and unexplained SCD, there was a 22% prevalence of a cardiac conduction disease as suggested by positive Ajmaline tests. This proportion was similar to the proportion in families with SCD at young age alone,7 although the average age of SCD was higher and the number of SCD cases was larger in families with premature atherosclerosis. Even among families with unexplained SCD (with or without premature atherosclerosis) at a relatively advanced age (>45 years), BrS was found in 16%. The underlying genetic basis for BrS differed between families with premature atherosclerosis and those without. While a SCN5A mutation was found in 18% of families with SCD alone (similar to reported proportions8 9), SCN5A mutations were virtually absent from families with SCD and premature atherosclerosis.
At present, screening for an underlying inherited disease is not conducted in families with unexplained SCD at older age, especially when premature atherosclerosis is present. Our finding that, in these families, BrS occurs at a similar prevalence as in families with SCD alone suggest that this strategy may need to be altered and that in these families BrS should be ruled out.
We can only speculate about the causes for the high proportion of positive Ajmaline tests in families with premature atherosclerosis and SCD.
First, accelerated atherosclerosis may cause changes in the myocardium that lead to conduction slowing, a key pathophysiological mechanism in BrS.10 Emerging evidence points to myocardial fibrosis as a particularly important factor.11 Atherosclerosis-induced fibrosis may facilitate the development of the hallmark ECG changes of BrS during Ajmaline testing and unmasking individuals with BrS. However, multiple young individuals from families with premature atherosclerosis in our study with a positive Ajmaline test had no calcified plaque on coronary CT scan.
Second, the double hit hypothesis, suggesting that families with both premature atherosclerosis and unexplained SCD have different, possibly multiple genetic, mutations leading to either conduction disease and/or premature atherosclerosis. If this would be the case, the general population prevalence of this arrhythmia must be much higher for it to appear at random among families with premature atherosclerosis. In addition, we observed that more than 30% of families with premature atherosclerosis also experience unexplained SCD.
Third, a common (genetic) cause may underlie both premature atherosclerosis and unexplained SCD.12 It was suggested that in many unexplained SCD cases, atherosclerosis and acute ischaemia would lead to arrhythmias due to a genetic modifier. For instance, factors leading to atherosclerosis, such as smoking, insulin resistance and high cholesterol, might also lead to damage of the sodium channels even before atherosclerosis can be observed.13 It is proposed that sodium channel defects are most likely a sliding SCD between distinct BrS and minor sodium channel defects, which in combination with mild ischaemia might lead to a deathly arrhythmia. Since we did not observe any cosegregation between a positive Ajmaline test and premature atherosclerosis, we could conclude that these two entities (genetic arrhythmia and premature atherosclerosis) can exist independently within a family. Therefore, our third hypothesis seems to most likely.
Strengths and limitations
With this paper, we substantiate our hypothesis that ischaemia-induced sodium channel dysfunction and conduction slowing may conspire with inherited reduction in cardiac excitability, such as BrS, to result in lethal arrhythmia and SCD. The best way to test this hypothesis would be a large prospective study of individuals of families with premature atherosclerosis either with or without SCD. All these individuals should undergo a thorough cardiac investigation, including ECG and Ajmaline testing; also, cardiac imaging (eg, echocardiography) should be performed to exclude structural abnormalities. Conducting such investigations for scientific reasons, however, would be ethically challenging in particular, because of the risk of arrhythmias during Ajmaline testing.14 Therefore, we decided to investigate the relationship between BrS and premature atherosclerosis in an exceptionally large cohort of individuals who underwent Ajmaline testing for clinical purposes (because of unexplained familial SCD). However, a limitation of this study design is the heterogeneity of the study population, which stems from disparate referral sources: individuals from families with both premature atherosclerosis and SCD (in which SCD cases were relatively old) and individuals from families referred from the cardiogenetics department because of familial SCD at a young age. Still when analysing the data without the families from the outpatient clinic of premature atherosclerosis, the results remained similar.
Conclusion
In families with unexplained SCD and premature atherosclerosis, the prevalence of a positive Ajmaline test was similar to that in families with unexplained SCD at young age alone without premature atherosclerosis. The underlying genetic defect is different between both groups, as SCN5A mutations are significantly less prevalent in families with premature atherosclerosis and SCD. Moreover, SCD in such families occurs at a relatively advanced age. Still these findings suggest that in families with premature atherosclerosis and familial SCD, screening for inherited diseases that increases vulnerability to SCD should be performed in particular, BrS.
Key messages
What is already known on this subject?
Generally, sudden cardiac death in families with premature atherosclerosis is attributed to a myocardial infarction.
What does this study add?
Our studies identified that sudden cardiac death in families with premature atherosclerosis could be related to an underlying genetic predisposition of arrhythmias.
How might this impact on clinical practice?
Our findings suggest that the sudden cardiac death in families with premature atherosclerosis is often falsely attributed to atherosclerosis and clinicians should consider family members to be tested for an underlying arrhythmia even when the sudden cardiac death occurs at older age. If patients are referred, one must keep in mind that the underlying genetic defect is different between both groups.
References
Footnotes
Contributors CB and MR contributed equally to this paper. MR and SP conceptualised and designed the study. CB, MR, AA and SJ contributed to analysis and interpretation. CB, MR, AA, SL, PL, UR, HT and SP performed the data collection. CB, MR and SJ drafted the paper. KH, AA, HT and SP revised the work and final approved the manuscript.
Funding HLT has received funding from the European Union's Horizon 2020 research and innovation programme under acronym ESCAPE-NET, registered under grant agreement No 733381.
Competing interests All authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.