Article Text

Abstract
Objective In patients with chronic coronary or peripheral artery disease enrolled in the Cardiovascular Outcomes for People Using Anticoagulation Strategies trial, randomised antithrombotic treatments were stopped after a median follow-up of 23 months because of benefits of the combination of rivaroxaban 2.5 mg two times per day and aspirin 100 mg once daily compared with aspirin 100 mg once daily. We assessed the effect of switching to non-study aspirin at the time of early stopping.
Methods Incident composite of myocardial infarction, stroke or cardiovascular death was estimated per 100 person-years (py) during randomised treatment (n=18 278) and after study treatment discontinuation to non-study aspirin (n=14 068).
Results During randomised treatment, the combination compared with aspirin reduced the composite (2.2 vs 2.9/100 py, HR: 0.76, 95% CI 0.66 to 0.86), stroke (0.5 vs 0.8/100 py, HR: 0.58, 95% CI 0.44 to 0.76) and cardiovascular death (0.9 vs 1.2/100 py, HR: 0.78, 95% CI 0.64 to 0.96). During 1.02 years after early stopping, participants originally randomised to the combination compared with those randomised to aspirin had similar rates of the composite (2.1 vs 2.0/100 py, HR: 1.08, 95% CI 0.84 to 1.39) and cardiovascular death (1.0 vs 0.8/100 py, HR: 1.26, 95% CI 0.85 to 1.86) but higher stroke rate (0.7 vs 0.4/100 py, HR: 1.74, 95% CI 1.05 to 2.87) including a significant increase in ischaemic stroke during the first 6 months after switching to non-study aspirin.
Conclusion Discontinuing study rivaroxaban and aspirin to non-study aspirin was associated with the loss of cardiovascular benefits and a stroke excess.
Trial registration number NCT01776424.
- coronary artery disease
- pharmacology
- clinical
- stroke
- acute coronary syndrome
- peripheral vascular diseases
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All the data for the present report came from the Population Health Institute (PHRI) in Hamilton, Canada. PHRI did also all data analysis for COMPASS. Data are available on reasonable request as outlined in the COMPASS data sharing policy described in the supplement.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- coronary artery disease
- pharmacology
- clinical
- stroke
- acute coronary syndrome
- peripheral vascular diseases
Introduction
Discontinuation of effective antithrombotic therapies such as aspirin in chronic coronary artery disease (CAD) or anticoagulants in atrial fibrillation is associated with increased risk of cardiovascular (CV) events due to the loss of their protective effect.1 2 In the Cardiovascular Outcomes for People Using Anticoagulation Strategies (COMPASS) trial involving 27 395 participants with chronic CAD or peripheral artery disease (PAD), the combination of low-dose rivaroxaban 2.5 mg two times per day and aspirin 100 mg once daily compared with aspirin 100 mg once daily reduced the risk of the primary outcome, a composite of myocardial infarction (MI), stroke or CV death by 24%, including a 42% reduction in stroke and a 22% reduction in CV death. Although the combination compared with aspirin alone increased major bleeding (3.1 vs 1.9%), the rate of the net benefit of the main outcome was reduced but remained statistically significant (4.7% vs 5.9%)3; it is noteworthy that the excess of major bleeding occurred during the first year after randomisation without statistically significant difference between the two randomised groups in the subsequent years.4 There was also a 47% reduction in risk of major adverse limb events and an 18% reduction in mortality.3 5 Rivaroxaban 5.0 mg two times per day compared with aspirin 100 mg once daily did not significantly reduce the primary outcome.3 The COMPASS trial was expected to last 3–4 years but was stopped early at the recommendation of the independent Data Safety Monitoring Board after a median follow-up of 23 months owing to evidence that the combination of low-dose rivaroxaban and aspirin was clearly beneficial. Given that the combined rivaroxaban 2.5 two times per day and low dose aspirin had not been approved for clinical use, it was necessary to obtain regulatory and ethics approvals before making the treatment available to study participants. Therefore, at the end of the trial, all participants were requested to switch to non-study low-dose aspirin once daily while awaiting regulatory approval.
We hypothesised that COMPASS trial participants who continued randomised antithrombotic treatment until the early stopping visit and discontinued study treatment with combination of rivaroxaban and aspirin to non-study aspirin compared with those who were randomised to aspirin would be at a greater CV risk. To verify this hypothesis, we assessed the rates of MI, stroke or CV death during randomised treatment and after switching to non-study aspirin at the time of early stopping of the antithrombotic arms of the trial.
Methods
Study design and population
The trial design, population and main results have been previously reported.3–7 In brief, COMPASS was a 3 by 2 partial factorial, multicentre, double-blind, randomised, placebo-controlled trial done in 27 395 men and women with chronic CAD or PAD. In addition to the randomised comparison between the combination of rivaroxaban 2.5 mg two times per day and aspirin 100 mg once daily or rivaroxaban 5.0 mg two times per day and aspirin 100 mg once daily, the COMPASS trial investigators randomised 17 598 of the 27 395 participants to either pantoprazole 40 mg once daily or matched placebo once daily. The 9117 participants randomised to rivaroxaban 5.0 mg two times per day were excluded from this report because this treatment did not reduce the primary outcome. The COMPASS study protocol had been approved by Ethics Committees at all participating sites, and all participants provided written informed consent.
Discontinuation of rivaroxaban and follow-up
In January 2017, the independent Data and Safety Monitoring Board recommended that the antithrombotic arms of the study be discontinued early because of clear benefit of the combination of rivaroxaban and aspirin compared with aspirin alone.8 This recommendation was accepted by the COMPASS Steering Committee and the sponsor on 6 February 2017. Between 6 February and 18 July 2017, all participants attended a close-out visit where they were informed of the study results, told to stop the rivaroxaban and aspirin trial medications and to switch to non-study aspirin at a dose of 75–100 mg once daily.
One month after the rivaroxaban/aspirin close-out visit, a questionnaire eliciting clinical events was administered by telephone to all participants. Participants enrolled in the proton pump inhibitor (PPI) arms of the COMPASS trial continued to attend study visits every 6 months as per protocol until August 2018. Finally, following regulatory agency and ethics approvals of the combination of rivaroxaban and aspirin in each country, all participants were invited to participate in the long-term open-label extension (LTOLE) study during which they would receive the combination of rivaroxaban 2.5 mg two times per day and low-dose aspirin once daily until such time that this regimen was commercially available or for a maximum of 3 years, whichever occurred first. All participants consenting to LTOLE underwent clinical re-evaluation prior to starting open-label rivaroxaban 2.5 mg two times per day plus aspirin 100 mg once daily. Thus, follow-up for the current analyses began at the time of early stopping of randomised antithrombotic treatments and continued until last contact, at the time of the 1 month questionnaire, the final PPI follow-up at the end of August 2018 or the start of the LTOLE open-label study, whichever occurred first (online supplemental figure 1). Because our focus was on the potential impact of switching to non-study aspirin at the time of early stopping of randomised antithrombotic treatments, we excluded from these analyses participants who had already discontinued antithrombotic study medications prior to the early stopping visit.
Supplemental material
Outcomes
The main outcome of interest was the composite of MI, stroke or CV death and its individual components, as well as acute limb ischaemia and limb amputation for vascular reasons. Outcomes were collected on standardised event forms with supportive documentation and were verified using a computer-programmed algorithm. A central adjudication committee reviewed and adjudicated the event if the algorithm did not confirm the event. Five events occurring while participants were on the non-study aspirin were received a few days after the LTOLE initiation visit and were accepted based on the diagnosis of the local principal investigators. All hospitalisations and revascularisations were recorded.
Statistical analyses
Categorical variables are reported as count and percentages and compared using Pearson χ2 tests. Continuous variables are reported as mean and SD or median and IQR and compared with two-sample t-test or Wilcoxon two-sample test as appropriate. All outcomes occurring from the time of early stopping of study antithrombotic treatments until last contact (at the time of the 1 month questionnaire, the final PPI follow-up at the end of August 2018 or the start of the LTOLE open-label study, whichever occurred first) were included in the analysis. Time-to-event was expressed in days with the first day corresponding to either the day of randomisation or the day of early stopping depending on the analysis and ending at time of an outcome event or the last contact. Incidence rates were estimated as the number of first events per 100 person-years (py). Kaplan-Meier estimates were used to estimate cumulative risks over time. Cox proportional hazard models (stratified according to PPI randomisation: not randomly assigned to a PPI, pantoprazole 40 mg, pantoprazole-matched placebo) were used to estimate HRs and corresponding 95% CIs, and comparisons between the combined rivaroxaban and aspirin group and the aspirin control group were performed by stratified log-rank tests. Landmark analyses were done for two time periods: time of early stopping to 6 months post early stopping and from 6 months post early stopping to the last contact post early stopping. Competing risk analyses of non-CV death for the composite outcome and CV death, and all-cause mortality for MI and stroke were done according to Fine-Gray subdistribution hazard models.9 The statistical software used was SAS V.9.4 (SAS Institute Inc, Cary, North Carolina, USA).
Results
Study population and baseline characteristics
Of the 18 278 randomised participants to the combination or aspirin alone followed during the COMPASS trial, 772 died, 2632 permanently discontinued the study medications because of events, side effects, procedural interventions or use of open-label anticoagulants, 46 withdrew their consent, 8 were lost to follow-up and 10 had no follow-up after the early stopping visit (online supplemental figure 2 and online supplemental table 1). Of the remaining 14 810 participants, the 14 068 who switched to non-study aspirin 75–100 mg once daily and had at least one contact after the early stopping visit constitute the study population: 7027 were in the group originally randomised to the combination of rivaroxaban and aspirin (4579 (65.2%) of these were followed every 6 months in the PPI study arm) and 7041 in the group originally randomised to aspirin alone (4539 (64.5%) of these were followed every 6 months in the PPI study arm). Table 1 describes their baseline characteristics at study entry. The mean age of the participants was 67.9 years and 22% were women. There were no significant differences in the clinical characteristics between the two study groups except for a slightly higher intake of non-steroid anti-inflammatory drug in the combination group.
Baseline characteristics by randomised treatment groups of participants who continued study antithrombotic treatments until early stopping and then switched to non-study aspirin
Outcomes during randomised treatment
As previously reported in the main results paper, the combination of rivaroxaban and aspirin compared with aspirin reduced MI, stroke or CV death by 24% (2.2 vs 2.9/100 py, HR: 0.76, 95% CI 0.66 to 0.86), stroke by 42% (0.5 vs 0.8/100 py, HR: 0.58, 95% CI 0.44 to 0.76) and CV death by 22% (0.9 vs 1.2/100 py, HR: 0.78, 95% CI 0.64 to 0.96), with no significant reduction in MI (1.0 vs 1.1/100 py, HR: 0.86, 95% CI 0.70 to 1.05).
Outcomes after switching to non-study aspirin
Figure 1 and table 2 present the outcomes in participants who continued taking study antithrombotic treatment until early stopping, switched to non-study aspirin and were followed until last contact, a median (25th and 75th percentile) of 1.02 (0.74 and 1.11) years. During this period, the composite outcome occurred in 240 participants: MI in 95, stroke in 66 (95% non-haemorrhagic strokes) and CV death in 102. Acute limb ischaemia occurred in 15 participants and limb amputation for vascular cause in nine participants. Participants originally randomised to the combination compared with those randomised to aspirin alone had similar rates of MI, stroke or CV death, MI and CV death but higher rates of stroke, although the ischaemic stroke excess did not reach statistical significance (table 2). Rates of acute limb ischaemia and amputation for vascular causes were similar. There was no difference in major bleeding between the two groups.

Outcomes from the time of switching to non-study aspirin until final contact in participants who took study antithrombotic drugs until early stopping (n=14 086). Panel a: composite outcome panel; panel B: cardiovascular death; panel C: MI; panel D: stroke. ASA, aspirin; MI, myocardial infarction; Riva, rivaroxaban.
Outcomes by randomised groups from time of switching to non-study aspirin to last contact in participants who took study antithrombotic treatments until the time of early stopping
During the first month after switching to non-study aspirin the event rates were very low; the composite outcome occurred in only 10 (0.1%) of the participants in the combined medication group and in 8 (0.1%) of the aspirin group. Most of the excess strokes in the participants in the combine treatment group occurred during the first 6 months (figure 2) including significant increase in ischaemic stroke (25 vs 7 events, 0.9 vs 0.2/100 py, HR: 3.55, 95% CI 1.54 to 8.21) (online supplemental table 2).
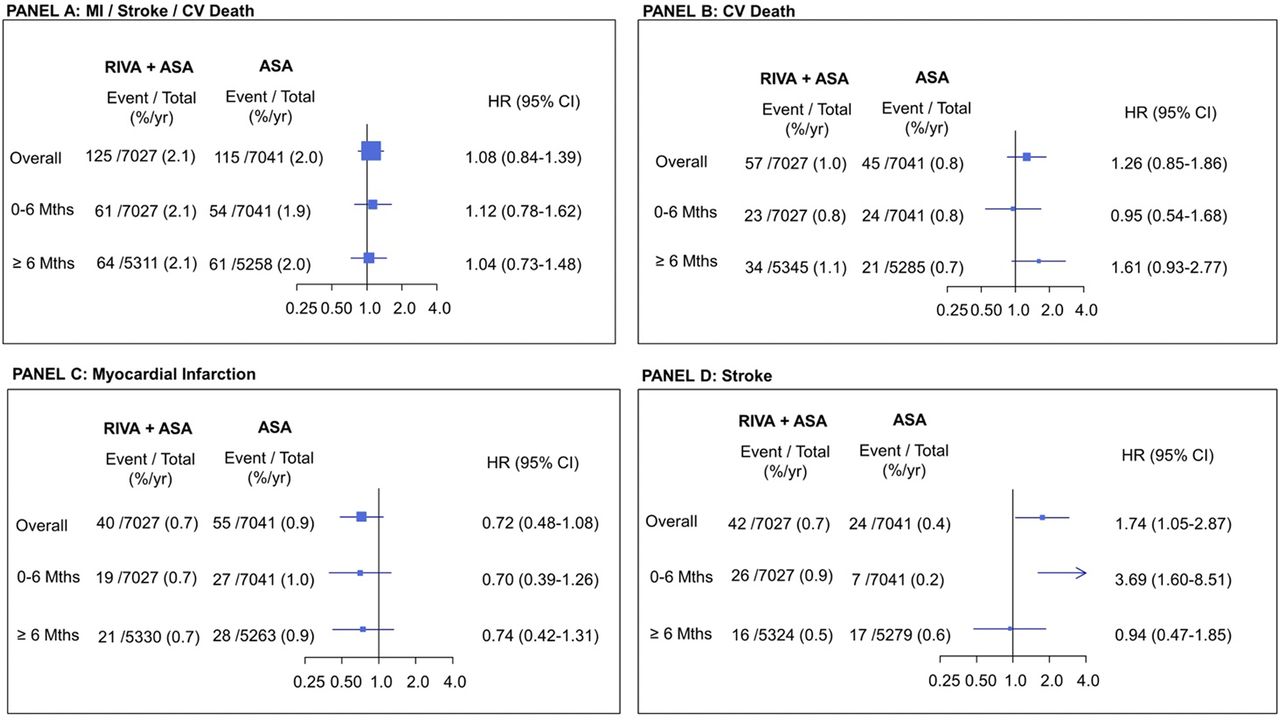
Landmark analysis: outcomes from the time of switching to non-study aspirin until final contact in participants who took study antithrombotic drugs until early stopping. Panel A: composite outcome; panel B: cardiovascular (CV) death; panel C: myocardial Infarction (MI); panel D: stroke. %/yr=per 100 person-years; ASA, aspirin; Riva, rivaroxaban.
Competing risk analyses of non-CV death for the composite outcome and CV death, and all-cause mortality for MI and stroke did not substantially change the estimates (online supplemental table 3).
Outcomes from randomisation to last contact
Figure 3 and table 3 show the outcomes occurring between randomisation and the last contact after early stopping of the antithrombotic arms of the trial, a median (25th and 75th percentile) follow-up of 2.89 (2.26 and 3.47) years. The composite outcome occurred in 1243 participants: MI in 518, stroke in 320 (90% non-haemorrhagic strokes) and CV death in 557. The combination of rivaroxaban and aspirin compared with aspirin reduced MI, stroke or CV death by 18% and stroke by 26%, with no difference in CV death or MI.

{kind=link}
{kind=link}
{kind=link}
Outcomes from randomisation until final contact after switching to non-study aspirin (n=18 276). ASA, aspirin; Riva, rivaroxaban.
Outcomes by randomised treatment groups from time of randomisation to final contact after switching to non-study aspirin at the time of early stopping
Discussion
This is the first report that examines the impact of discontinuation of the combination therapy with rivaroxaban 2.5 mg two times per day and aspirin 100 mg once daily and switching to low-dose aspirin on risk of subsequent CV events in patients with chronic CAD or PAD. Discontinuing the combination of low-dose rivaroxaban and aspirin and switching to non-study aspirin was associated with a loss of the benefits of the combination and an excess of stroke particularly during the first 6 months after switching.
The present analysis was not prespecified as we had not anticipated early stopping of the trial for benefit. We acknowledge that participants who continued randomised antithrombotic treatments until the time of early stopping and who were switched to non-study aspirin were at lower risk than those originally randomised even though their baseline characteristics by treatment group were similar (online supplemental table 4). However, this is likely to lead to an underestimate of the potential harms of discontinuing the combination for non-study aspirin. Finally, we could not exclude an effect of confounding for example due to subclinical atrial fibrillation or changes in other risk factors over time. However, since the baseline characteristics of treatment groups participating in the follow-up after stopping rivaroxaban were well balanced, it is unlikely that changes in risk factors and subsequent cointerventions differed by treatment group.
It is unclear why event rates after switching to non-study aspirin were low during the first month, and the excess of stroke did not become evident until 2–6 months after switching. This finding contrasts with prior reports of an early excess of ischaemic events after stopping anticoagulant therapy.10–13 One possible explanation is that prior reports were mostly from trials of patients with atrial fibrillation who are prone to immediate thrombus formation in the left atrial appendage when antithrombotic therapy is stopped. Furthermore, in these earlier trials, there was relatively little use of aspirin that may exert longer term protective effects. An analysis from the Swedish national registry demonstrated that discontinuing low-dose aspirin (75–160 mg) was associated with an increase in ischaemic events,1 but similar to COMPASS, there was no strong temporal relationship between aspirin discontinuation and the occurrence of ischaemic events. Nevertheless, the Swedish study results support previous work showing that interruption of low-dose aspirin for more than 24 hours was associated with increased CV risk.14 In the COMPASS trial, aspirin therapy was not interrupted because all participants switched from the study antithrombotic therapy (the combination rivaroxaban and aspirin) and the study aspirin to non-study aspirin. Our results suggest that when low-dose aspirin is maintained, the excess absolute risks of events after stopping rivaroxaban for a short period (eg, 1–2 weeks) is low.
Compared with the randomised treatment period in COMPASS trial (median 1.88 years), the benefits of the combination treatment compared with aspirin in reducing the primary composite, MI, stroke or CV death, were attenuated when taking into account the entire follow-up period between randomisation and last contact after early stopping of study antithrombotic treatments and switching to non-study aspirin (median 2.89 years). This observation is consistent with the finding of no additional benefit after rivaroxaban is discontinued. Although disease progression could lead to attenuation of benefit independently of whether randomised treatment is stopped, this cannot explain our results. First, the lack of continued benefit was abrupt, coincided with switching to non-study aspirin. Second, as observed with anticoagulant therapy discontinuation, the lack of continued benefit was independent of both the duration of randomised treatment prior to switching and disease severity. Third, disease progression would be expected to be similar in both arms, whereas in our study, there was an excess of stroke in participants who switched from the combination to non-study aspirin alone. Fourth, prior studies have shown that benefits of antithrombotic therapy in patients with vascular disease continue to accrue over many years without attenuation.15 16 Finally, our results are consistent with prior reports of harm after stopping effective antithrombotic therapies.2
The combination of low-dose rivaroxaban and aspirin reduces the risk of atherothrombotic vascular events by targeting coagulation factor Xa and platelet cyclo-oxygenase-1, respectively. In addition to promoting thrombin generation, factor Xa interacts with protease-activated receptors 1 and 2 that may activate platelets and increase endothelial dysfunction and inflammation contributing to the atherothrombotic process.17 The reduction of stroke in the COMPASS trial with the combination of rivaroxaban and aspirin compared with aspirin (HR: 0.58, 95% CI 0.44 to 0.76) illustrates a pronounced reduction in thromboembolic events.18
Considering the pronounced stroke reduction in the trial, discontinuation of the combination and switching to non-study aspirin exposed the participants to a greater risk for this event, confirming our hypothesis. Switching was not associated with a significant increase in CV death, but there was a loss of benefit as also seen for the primary outcome. The reason for the lack of a significant change in MI is unclear but suggests that this outcome is less sensitive to discontinuation of rivaroxaban than stroke.
Implications
Our results demonstrating adverse consequences of discontinuing efficacious trial medication at the end of the trial while waiting for regulatory agencies or ethics board approvals have important implications for future conduct of clinical trials. Although some trials have built into their protocol a statement that efficacious trial medication should be available for the participants when the trial is terminated, this is not the case for most trials. Furthermore, even when prespecified in the protocol, provision of efficacious medications at the end of a trial and particularly if the trial is stopped early can pose logistic, regulatory and ethical challenges and may not be feasible. Nevertheless, the potential for harm as demonstrated in our study mandates efforts to overcome these challenges and thereby uphold the best interests of trial participants. We believe that guidelines should be developed to enable continued use of efficacious treatments for trial participants until regulatory agencies more formally evaluate such therapies for routine clinical use and without requiring the need for approval from individual ethics committees in multiple countries and sites.19 20
Conclusion
Our study suggests that in people with chronic CAD or PAD, discontinuation of rivaroxaban 2.5 mg two times per day and aspirin 100 mg once daily and switching to non-study aspirin was associated with a loss of benefits of the combination and an excess of stroke. Our results highlight that extended interruption or discontinuation of low-dose rivaroxaban in people with chronic CAD or PAD should be avoided, even if low dose aspirin is maintained, unless there is major medical reason. Our results also raise concern regarding the absence of post-trial access of participants to therapies shown to be efficacious during the trial.
Key messages
What is already known on this subject?
The Cardiovascular Outcomes for People Using Anticoagulation Strategies trial showed that the combination of rivaroxaban 2.5 mg two times per day and aspirin 100 mg once daily compared with aspirin 100 mg once daily reduced cardiovascular events in participants with chronic coronary or peripheral artery disease. However, the cardiovascular consequences of discontinuing the combination to non-study aspirin while waiting for the approval for clinical use of the combination by the regulatory agencies are unknown.
What might this study add?
In these participants, switching low-dose rivaroxaban and aspirin to aspirin at the study end was associated with a rapid loss of the benefits of the combination and an increased risk of stroke particularly between the second and sixth month after stopping the study medication. Discontinuation of efficacious study medication while waiting for approval exposed participating patients to potential harm.
How might this impact on clinical practice?
Adherence to the study combination is clinically important. The study suggests if low-dose rivaroxaban is discontinued for medical reason and aspirin is maintained the absolute risk of events is low for the first 2 weeks since the stroke risk occurred after the first month. To reduce the adverse consequences of discontinuing efficacious trial medication at the end of the trial while waiting for regulatory approvals, access by participants to efficacious therapies should be available.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All the data for the present report came from the Population Health Institute (PHRI) in Hamilton, Canada. PHRI did also all data analysis for COMPASS. Data are available on reasonable request as outlined in the COMPASS data sharing policy described in the supplement.
Ethics statements
Ethics approval
This study is a secondary analysis of the COMPASS trial (Clinical Trial Registration: URL: https://www.clinicaltrials.gov Unique identifier: NCT01776424). Study design and results of the COMPASS trial have been published, and the main publications are identified in the references of the present study as: 3, 4, 5 and 6. The COMPASS trial protocol was approved by each site regional ethics committee, and all participants gave written informed consent.
Acknowledgments
The authors would like to thank the participants and the staff of the Cardiovascular Outcomes for People Using Anticoagulation Strategies (COMPASS) trial for their valuable contributions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Collaborators N/A.
Contributors GRD and JWE design the substudy and wrote the first version; LD did the statistical analyses. All authors were involved in the COMPASS trial. They reviewed the data and provided comments on the different versions. All authors approved the final version of the manuscript.
Funding The COMPASS study was sponsored by Bayer AG, Germany.
Competing interests GRD reports speaking honoraria from Bayer and Eli-Lilly. JB serves on the Advisory Board of and has received honoraria from Bayer AG. DL has received speaking honoraria from Bayer AG. VA received honoraria from Bayer, Amgen, NovoNordisk, Novartis, BMS/Pfizer alliance and Sanofi. SDB is employed by Bayer U.S., LLC, as a clinical research physician. DLB reports the following relationships advisory board: Cardax, Cereno Scientific, Elsevier Practice Update Cardiology, Medscape Cardiology, PhaseBio, PLX Pharma and Regado Biosciences; board of directors: Boston VA Research Institute, Society of Cardiovascular Patient Care and TobeSoft; chair: American Heart Association Quality Oversight Committee; Data Monitoring Committees: Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute, for the PORTICO trial, funded by St. Jude Medical, now Abbott), Cleveland Clinic (including for the ExCEED trial, funded by Edwards), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo), Population Health Research Institute; honoraria: American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Vice-Chair, ACC Accreditation Committee), Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute; RE-DUAL PCI clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor in Chief, Harvard Heart Letter), Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (editor in chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (guest editor; associate editor), Medtelligence/ReachMD (CME steering committees), Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee and USA national coleader, funded by Bayer), Slack Publications (Chief Medical Editor, Cardiology Today’s Intervention), Society of Cardiovascular Patient Care (secretary/treasurer), WebMD (CME steering committees); other: Clinical Cardiology (deputy editor), NCDR-ACTION Registry Steering Committee (chair), VA CART Research and Publications Committee (chair); research funding: Abbott, Afimmune, Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Cardax, Chiesi, CSL Behring, Eisai, Ethicon, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Idorsia, Ironwood, Ischemix, Lexicon, Lilly, Medtronic, Pfizer, PhaseBio, PLx Pharma, Regeneron, Roche, Sanofi Aventis, Synaptic and The Medicines Company; royalties: Elsevier (Editor, Cardiovascular Intervention: A Companion to Braunwald’s Heart Disease); site coinvestigator: Biotronik, Boston Scientific, CSI, St. Jude Medical (now Abbott) and Svelte; trustee: American College of Cardiology; unfunded research: Flow Co, Merck, Novo Nordisk and Takeda. SJC has received major research grants, consulting fees and speaker fees from Sanofi-Aventis, Janssen, Pfizer, Bristol-Myers Squibb, Boehringer-Ingelheim, Boston Scientific, Abbott, Bayer Pharmaceuticals Inc, Portola Pharmaceutical, Medtronic, Daiichi Sankyo and Servier. KAAF has received grants from Bayer/Janssen and AstraZeneca and has received consultancy fees from Bayer/Janssen, Sanofi/Regeneron and Verseon. EM is employed by Bayer AG, as a global clinical leader. PW has received grants and honoraria from Bayer AG, Boehringer Ingelheim, AstraZeneca and Servier. BRW has received speaking honoraria from Bayer AG. SY has received grants and honoraria from Bayer AG, Boehringer-Ingelheim, AstraZeneca, Bristol-Myers Squibb and Cadila Pharmaceuticals. JWE has received consulting fees and grant support from AstraZeneca, Bayer AG, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi-Sankyo, Eli Lilly, GlaxoSmithKline, Pfizer, Janssen, Sanofi and Servier.
Provenance and peer review Not commissioned; internally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.