Article Text

Abstract
Sudden arrhythmic death syndrome (SADS) accounts for approximately 500 deaths in England and Wales per year. Clinical screening of the surviving first-degree relatives can identify an inherited cardiovascular condition in up to half of families, permitting lifestyle modification and confirmed effective prophylactic therapies to prevent further sudden deaths. Mechanisms for molecular autopsy are available to improve the diagnostic yield but practical barriers to its successful implementation exist. This article reviews the clinical screening of the first-degree relatives of SADS patients, molecular autopsy of probands and the broader implications of national recommendations for the investigation of sudden cardiac death.
- Cardiac channelopathies
- cardiomyopathies
- coroners
- genetic testing
- genetics
- sudden adult death syndrome
- sudden cardiac death
Statistics from Altmetric.com
- Cardiac channelopathies
- cardiomyopathies
- coroners
- genetic testing
- genetics
- sudden adult death syndrome
- sudden cardiac death
Four hundred and ninety-one thousand three hundred and forty-eight deaths were registered in England and Wales in 2009.1 Sudden cardiac death is responsible for approximately 60 000 deaths per year in the UK, with ischaemic heart disease the predominant cause, increasing with age.2 Table 1 lists the causes of sudden cardiac death. Sudden arrhythmic death syndrome (SADS) is defined as sudden unexpected death at the age of 4 to 64 years, with no previous cardiac history, last seen alive within 12 h of being found dead and with no cause identifiable on postmortem examination (including negative toxicology and ideally an expert cardiac pathologist review).3 Prospective studies of sudden unexplained cardiac death in apparently healthy Caucasians have estimated that SADS is responsible for between 143 and 544 deaths per year in England.3 4 A lower age limit of 1 year is now recognised and differentiates from sudden infant death syndrome, which comprises different aetiologies, although inherited arrhythmia syndromes do contribute to at least 10% of cases.5
Causes of sudden cardiac death
Cardiac channelopathies (long QT syndrome (LQTS), Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia) and cardiomyopathies (hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy) are diagnosed in up to half of families affected by SADS.6–9 There is a significant prevalence of disease in families with inherited cardiomyopathies and channelopathies, in which the mutation did not occur de novo in the proband, because the majority are inherited in an autosomal dominant pattern with a 50% likelihood of each first-degree relative being affected.10 The first-degree relatives of SADS patients are recommended to have clinical screening to determine if any other family members are affected and require monitoring or appropriate intervention.
The role of the SADS clinic
A quality requirement in chapter 8 of the National Service Framework mandates that ‘NHS services have systems in place to identify family members at risk and provide personally tailored, sensitive and expert support, diagnosis, treatment, information and advice to close relatives.’11 The recommendation is that families should be assessed in a dedicated clinic with appropriately trained staff, and figure 1 illustrates the sequence of screening processes undertaken. A specialist nurse is advisable to coordinate the clinic and outpatient testing, act as a point of contact for families, liaise with the coroner's office to obtain a copy of the postmortem report and enquire regarding the availability of retained tissue for possible molecular autopsy.
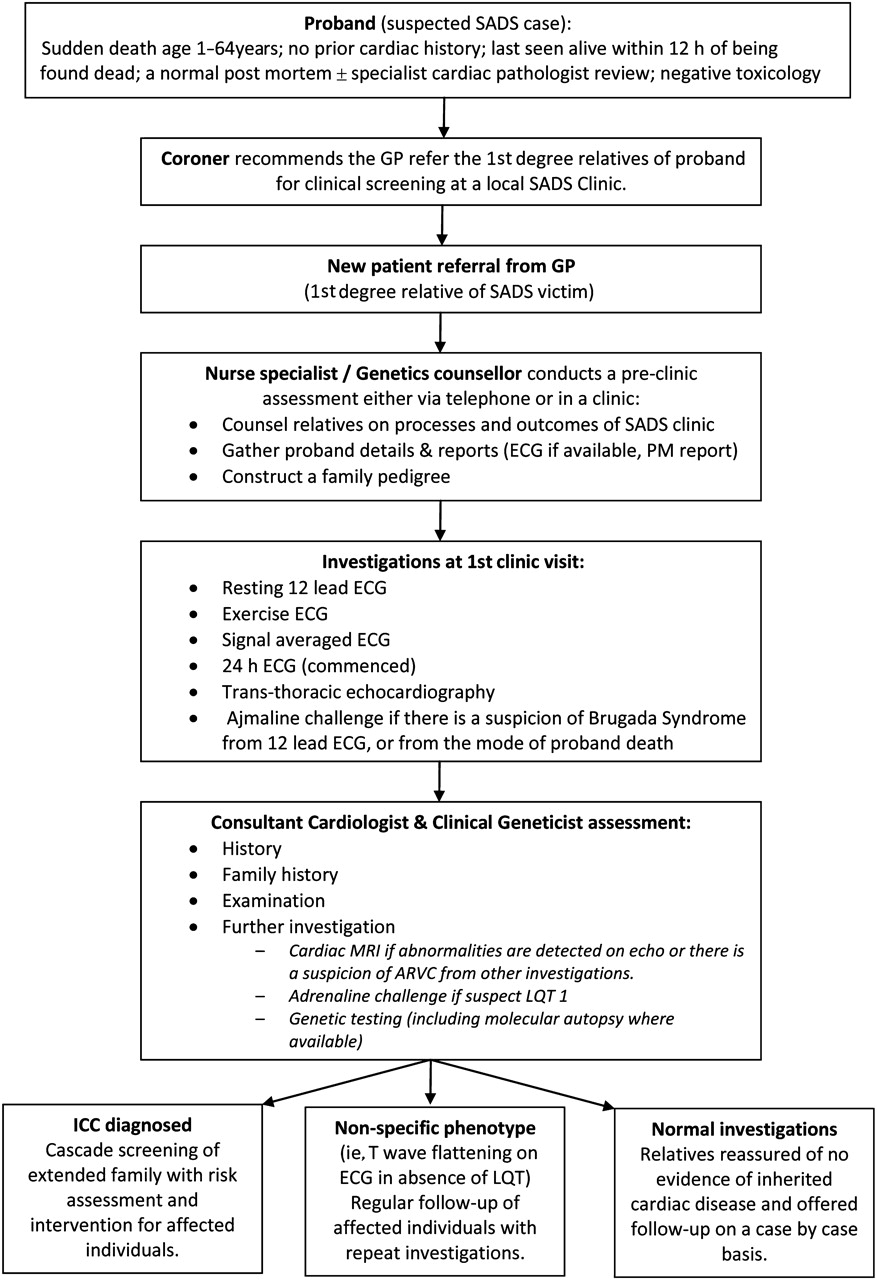
Clinical pathway for new referrals to the sudden arrhythmic death syndrome (SADS) clinic. ARVC, arrhythmogenic right ventricular cardiomyopathy; GP, general practitioner; LQT, long QT; ICC, inherited cardiac condition.
It is imperative that families are counselled regarding the possible repercussions of the testing, as a diagnosis of an inherited cardiac condition could have a significant impact on their ability to obtain insurance, employment, their psychological wellbeing and that of relatives and children.12 Relatives can be bombarded with several screening tests, with outcomes varying from inconclusive non-diagnostic findings necessitating further testing to ascertain their significance and regular follow-up, to medical therapy (ie, beta-blockers for LQTS) or even an implantable cardioverter defibrillator (ICD). Collaboration is required with a paediatric cardiologist for relatives with young children who will require screening and cardiologists with expertise in the diagnosis and management of cardiomyopathy because phenotypes can be difficult to characterise.12
The process begins with a detailed evaluation of the proband, the key features of which are:
Medication history (prescribed, over the counter and recreational) to screen for potential QT prolonging agents.
All potential sources of a premorbid ECG should be explored (ie, attendance at the emergency department, preoperative assessment, general practice, insurance medical) to examine for the features of a channelopathy or cardiomyopathy.
Circumstances of death can provide a clue to aetiology as certain triggers are well established (swimming/exertion in LQT 1, auditory triggers/during postpartum period in LQT 2 and sleep/rest in LQT 313 and in Brugada syndrome).14
Postmortem report including an expert cardiac pathologist review if possible.
The family pedigree is reviewed to identify any possible symptomatic relatives who may be affected and the instances of other sudden deaths in the family. Key features to elicit are:
A diagnosis of epilepsy should be reviewed especially if there are any atypical features in the history or a failure to respond to antiepileptic medication as misdiagnosis has been reported.15
An unexplained drowning or road traffic accident can alert to the possibility of an arrhythmia syndrome.
Relevant comorbidities can also provide diagnostic clues such as a premature requirement for pacemakers in conduction disease associated with SCN5A16 or Lamin A/C mutations.17
First-degree relatives undergo a thorough clinical assessment as outlined in figure 1 to unmask the phenotype of an inherited cardiac condition. Symptoms of syncope in a first-degree relative, two or more sudden unexplained deaths in a family and younger SADS patients are predictors of establishing a diagnosis in a family; however, associations are not consistent across the published studies.7–9 An essential component of the assessment is pharmacological testing, which is particularly important in the context of Brugada syndrome (ajmaline/flecainide challenge test) and may be helpful in certain forms of LQTS (epinephrine challenge) (figure 2).

{kind=link}
{kind=link}
Pharmacological testing. (i) Ajmaline challenge test demonstrating type 1 coved ST elevation in a sudden arrhythmic death syndrome (SADS) relative whose son died during sleep indicative of a Brugada syndrome phenotype in the family. (ii) Development of marked QT prolongation and bifid T waves in a patient carrying a KCNQ1 mutation causing long QT type 1.
The clinical screening process is employed in the first instance with confirmatory genetic testing to facilitate cascade screening of the wider family. The low diagnostic yield of initial genetic screening alone in the context of a normal phenotype makes this approach financially untenable.18 The main benefits of genetic testing arise from facilitating targeted therapy that impacts on prognosis, influences risk stratification or reproductive counselling. A recent Heart Rhythm UK consensus document outlined the role of gene testing in the conditions relevant to SADS screening.19
Having established a diagnosis, the prevention of further deaths in the family is achievable. Relatives are counselled on appropriate lifestyle measures and the avoidance of known triggers. Lists of drugs that should be avoided or used with caution in LQTS and Brugada syndrome are available and are continually updated online (http://www.azcert.org/ and http://www.brugadadrugs.org/). Both LQTS and catecholaminergic polymorphic ventricular tachycardia can be effectively managed by beta-blockade or other antiadrenergic measures (ie, left cervical sympathectomy). For example, beta-blockers alone reduce death rates from 78% to 6% in LQTS.20 The development of the ICD has provided a further highly effective measure in inherited arrhythmia syndromes and a Heart Rhythm UK position statement for ICD therapy has recently been published.21
SADS clinic diagnostic yield
Screening of the first-degree relatives in specialist clinics can diagnose an inherited cardiac condition in 22–53% of families. Behr et al6 investigated 109 first-degree relatives of 32 people who died of SADS. Seven (22%) of the 32 families were diagnosed with an inherited cardiac disease. A subsequent more detailed evaluation of 57 consecutively referred families with a SADS death identified 30 families (53%) with an inheritable heart disease.8 Over half of those relatives affected received a potentially lifesaving intervention with beta-blockers and/or an ICD. Tan et al7 investigated 43 consecutive families with one or more sudden unexplained death patient who died at less than 40 years of age. Only 51% of probands underwent a postmortem. One hundred and eighty-three relatives were initially screened, triggering the cascade screening of a further 150 relatives. A diagnosis was established in 17 of 43 families (40%) and revealed 151 presymptomatic disease carriers. Most recently, van der Werf et al9 reported the assessment of 140 families with a sudden unexplained death (aged 1–50 years). A diagnosis was established in 33% of families although a postmortem was performed in only 46% of cases. Strategies to increase the diagnostic yield include: ensuring all cases have a postmortem, ideally with an expert cardiac pathologist review of the whole heart; reviewing as many first-degree relatives in each family as possible; and carrying out a molecular autopsy when possible.
Molecular autopsy
Molecular autopsy, the postmortem genetic testing for inherited cardiovascular conditions known to cause SADS, has the promise of increased diagnostic yield and facilitating the cascade screening of relatives with more focused testing and the use of resources rather than the broad approach of current clinical screening protocols. DNA is extracted from retained tissues remote (months even years) from the postmortem. DNA can be used for confirmatory testing of a mutation identified in a surviving relative, or if sufficient good quality DNA is available, a comprehensive testing protocol for SADS can be employed. The first reported case of an inherited arrhythmia syndrome diagnosed by molecular autopsy was published in 1999.22 Subsequent to this a number of reports have been published of molecular autopsy in SADS cases.6 8 23–30 These are summarised in table 2. This process identifies a known mutation or possible disease-causing mutation in 0–35% cases and thus is best utilised in concert with the clinical screening of relatives.
Published evidence for molecular autopsy
The most commonly available archived tissue to extract DNA from is in the form of formalin-fixed paraffin-embedded tissue. This is stored at room temperature and is transported easily. The disadvantage is that DNA extraction from this source can generate novel false-positive sequence variations and does not permit the comprehensive interrogation of all exons in a large gene such as the cardiac ryanodine receptor gene (RYR2) implicated in catecholaminergic polymorphic ventricular tachycardia.31 It is possible to use this DNA for confirmatory testing for a mutation already identified in family members. EDTA-preserved blood kept at −20°C in an ordinary freezer and fresh frozen tissue particularly spleen kept at −80°C permit the extraction of sufficient (approximately 10 μg) good quality DNA to enable next generation sequencing and more comprehensive genetic assessment. This necessitates having liquid nitrogen available and access to specialist freezers, resources that are not commonly available. An alternative is RNAlater solution, which can be used to preserve RNA at room temperature without the need for immediate freezing, and genomic DNA can then be successfully reverse sequenced later from the preserved RNA.
Feasibility of molecular autopsy
Despite the evidence for molecular autopsy and ample guidance from the Association for European Cardiovascular Pathology,32 the Department of Health11 and Heart Rhythm UK19 for it to be performed as part of the investigation of sudden cardiac death, it is underutilised. Genetic testing of the proband was reported in only 10% of SADS patients in recent publication of SADS clinics.6–9 With advances in technology and the wider availability of genetic testing in clinical practice the flow-limiting step is the availability of proband DNA.
The Human Tissue Act (2004) is the primary legislation that deals with the use and storage of postmortem samples and related issues of consent in England and Wales. This legislation was put before parliament in the wake of public enquiries carried out into the inappropriate removal and storage of human tissue at Bristol Royal Infirmary and Alder Hey hospitals. As with any piece of legislation, only a court can definitely interpret its meaning. The dearth of litigation in this area, which might provide detailed and definitive guidance, leaves a situation in which different people may interpret the Act in different ways in daily practice. Guidance is available from both the British Medical Association and the Department of Health (see appendix 1). Samples removed under the authority of a coroner for the purpose of establishing cause of death are exempt from the Human Tissue Act. This will not apply to allow the removal of samples once a coroner has discharged their duty, as samples cannot be retained or used to obtain information ‘which may be relevant to any other person (including a future person) without relatives consent’. It is common for relatives to state later in clinic that they would have consented to tissue retention if approached at the time. Concerns over possible litigation, lack of appropriate storage facilities for retained tissues in mortuaries or lack of access to counselling to discuss tissue retention with a view to genetic testing with the family at a difficult time all contribute to missed opportunities.
The role of the coroner
Coroners play a critical role here, for it is under their authority that a postmortem is conducted in cases of sudden death. To appreciate their part in this requires knowledge of the coronial system, a short explanation of which follows: Coroners are independent judicial officers funded by the relevant local authority in whose territory they operate. They are lawyers/doctors of at least 5 years' practice and there are approximately 112 currently in England and Wales. Their primary function is to ascertain the cause of death in certain types of cases, including those in which the cause of death is unknown or is sudden and unexplained. A coroner is not part of the NHS and as such has no statutory duty of care to the surviving relatives. There is a duty to prevent further deaths in their district, which some coroners interpret to include sanctioning the retention of tissue for the purpose of genetic testing for SADS patients, whose relatives are potentially at risk of an inherited condition that may be fatal.
Two hundred and twenty-nine thousand eight hundred and eighty-three deaths (47% of all deaths registered) were reported to coroners in 2009, resulting in 105 354 postmortem examinations (46% of all deaths reported to coroners).33 If the pathologist is able to identify a cause of death that is neither violent nor unnatural then they will certify it to the coroner who will be able to issue the necessary documents for the death to be registered. At this point the coroner has discharged their duty. Any tissue samples retained from the postmortem are then no longer exempt from the Human Tissue Act, and without the explicit consent of the family samples are returned for burial, cremation or are destroyed. This occurs before relatives are referred to the SADS clinic and have access to genetic counselling. There must be clear lines of communication between the coroner's office and pathologists so that in cases of SADS the pathologist can act under the authority of the coroner to permit the retention of the whole heart for an expert cardiac pathologist review and retain appropriate tissue samples to help establish the cause of death and ensure that the opportunity for a molecular autopsy is not lost.
Pathologists receive a standard fee of £97 for conducting and reporting a postmortem regardless of how long it takes or the level of detailed examination required. Time pressures and a lack of resources can deter some pathologists from undertaking coronial cases. The National Confidential Enquiry into Patient Outcome and Death (NCEPOD) report 2006 has recommended independent audit of the quality and detail of the coroner's autopsy examination and report.34 The UK Cardiac Pathology Network provides a list of pathologists with a specialist interest in cardiac pathology who can provide an expert referral service for coroners and links to training courses and initiatives in cardiac pathology. DNA extraction and testing does not need to occur immediately, rather tissue samples just need to be extracted and retained under the coroner's authority while the family are counselled and approached for consent to retain the samples beyond death certification under the authority of the Human Tissue Act. The opportunity is then not lost for the SADS clinic to undertake a molecular autopsy in due course.
The broader implications of National Service Framework chapter 8 implementation
The creation of a SADS clinic service to diagnose and manage SADS families is a technically demanding process requiring considerable input from genetic counsellors, specialist nurses, clinical geneticists, electrophysiologists and cardiologists with expertise in cardiomyopathy and cardiac imaging. With the evolution of genetic testing, access to genetic testing facilities is extremely useful for some conditions to make a diagnosis and guide management.21 Screening should be undertaken by those with experience in managing inherited cardiovascular conditions as these conditions are characterised by significant genetic heterogeneity (>400 mutations have been identified in 12 LQTS susceptibility genes)13 and incomplete penetrance (identical pathogenic mutations can result in a spectrum of clinical severity in different individuals). This is further complicated by the fact that even in the absence of a typical phenotype, gene carriers can be at significant risk of cardiac events. In a recent study long QT mutation carriers with a normal corrected QT interval were still found to have a 10-fold greater risk of events compared with mutation-negative relatives.35 This molecular approach does have its pitfalls, as previously undescribed mutations may be identified. These may be pro-arrhythmic but require evaluation of gene carriers and protein modelling studies to establish their clinical relevance. This uncertainty is also compounded by the fact that other polymorphisms can modify the severity of a given phenotype altering the pro-arrhythmic potential of that particular mutation in a given family member.36 37 These factors influence the repolarisation reserve in LQTS and can create a normal QTc on a mutation carrier's ECG. The identification of an ion channel mutation in a family does not necessarily mean that this mutation is disease causing in a given family member, as non-allelic polymorphisms of that gene may also be protective. Therefore, careful evaluation of all the clinical data is required to formulate a management plan in any given family. In the future, it may be possible to evaluate the entire ‘electrical gene profile’ to assess the overall risk, but this will require a number of years of meticulous cellular and whole organ-based cardiac research to evaluate the impact of the most common ion channel mutations, polymorphisms and their interactions. In the meantime, optimal implementation of chapter 8 requires that a network of SADS clinics is established nationally. This is likely to number approximately 10 outside London,38 with close collaboration between clinics essential to ensure the prompt sharing of clinical information to facilitate the clinical screening of families with relatives living in different regions. The clinical expertise and systems put in place will also be responsible for managing all families with inherited cardiac conditions and the survivors of idiopathic ventricular fibrillation and their families.
There need to be clear lines of communication between coroners and pathologists to ensure that a detailed standardised SADS-specific postmortem is performed and an appropriate collection and storage of cardiac tissue and tissue for DNA extraction. Defined referral pathways from the coroner to the family general practitioner (GP) and SADS service need to be established with the GP acting as the channel to facilitate the referral. This requires education of both coroners and GPs to make them aware of the appropriate local SADS specialist service. Furthermore, central registration of SADS deaths would facilitate resource allocation and provide vital epidemiological information to plan service provision. Although this register exists,39 there are issues with its maintenance in the current economic climate. Closer liaison between the coronial system and the medical services available would ensure that families are referred appropriately and promptly to a SADS specialist clinic and maximise the utilisation of resources available. These challenges are not unique to this country and although there is some variation between countries in the precise legal framework in which SADS services operate, these are universal issues faced by all. A recent European survey40 highlighted the disconnect between forensic services (ie, coroners, procurator fiscals, magistrates and pathologists) who investigate sudden death but have no duty of care to the family of the person who has died and the clinicians (cardiologists and geneticists) who undertake the clinical screening of surviving relatives. The expertise is in place but we need to ensure that there is flow of information between the investigating parties. In the USA such networks have not been developed to date and referrals are made by family practitioners to a local cardiologist or specialist with an interest in inherited cardiovascular disease. There is no systematic national referral protocol for SADS family members. Referral itself is often dependent upon the family members wishing to be screened or on the recommendation of the family practitioner, particularly if the coroner has diagnosed a cardiomyopathy. Referral for investigation is also affected by the level of medical insurance cover relatives have as to whether investigations to exclude inherited disorders will be remunerated. In some institutions investigations may be completed using charitable funds. The family screening protocols employed are also not standardised, although most physicians employ a physical examination, ECG and a Holter if there is a suspicion of conduction disease or LQTS. A family history suggestive of structural heart disease/cardiomyopathy would prompt detailed imaging with echocardiography with or without MRI. There currently are no national guidelines for family screening in SADS in the USA.
The challenge over the next decade is to ensure the routine retention of suitable tissues for optimal DNA extraction, with the facilities to store them and provide combined genetic counselling with expert clinical screening in a fully integrated SADS family evaluation service throughout the UK.
Acknowledgments
The authors would like to thank the following: Dr Daniel Jacoby and Dr Mark Schoenfeld, Yale University School of Medicine, USA for their insight into family screening in the USA; Dr Henning Bundgaard, National University Hospital of Copenhagen, Denmark; Dr Juan R Gimeno, University Hospital Virgen de la Arrixaca, Murcia, Spain; Dr Victoria Murday, Yorkhill Hospital, Glasgow, Scotland; Dr Deirdre Ward, Adelaide & Meath Hospital, Dublin, Ireland; Dr Esther Zorio, Hospital La Fe, Valencia, Spain for their insight into family screening in Europe.
Appendix
Human Tissue Legislation. Guidance from the BMA Medical Ethics Department. (12/01/2011); available from: http://www.bma.org.uk/images/humantissuelegislationaug2009_tcm41-190148.pdf
The Human Tissue Act 2004. New legislation on human organs and tissue. (12/01/2011); available from:http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_4109591.pdf
Human Tissue Act 2004. (18/01/2011); available from: http://www.legislation.gov.uk/ukpga/2004/30/contents
References
Footnotes
Funding This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. LMN has received an educational grant from St Jude Medical.
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.