Article Text

Abstract
Cardiac amyloidosis of transthyretin fibril protein (ATTR) type is an infiltrative cardiomyopathy characterised by ventricular wall thickening and diastolic heart failure. Increased access to cardiovascular magnetic resonance imaging has led to a marked increase in referrals to our centre of Caucasian patients with wild-type ATTR (senile systemic) amyloidosis and Afro-Caribbean patients with the hereditary ATTR V122I type. Both subtypes present predominantly as isolated cardiomyopathy. The differential diagnosis includes cardiac amyloid light-chain (AL) amyloidosis, which has a poorer prognosis and can be amenable to chemotherapy. We review here the clinical features of cardiac ATTR amyloidosis and describe the diagnostic tests to determine ATTR type. Correct diagnosis is ever more crucial given that several novel therapies for ATTR amyloidosis are on the near horizon.
- Cardiac amyloidosis
- cardiovascular MRI
- cardiomyopathy
- valvular disease
- cardiac function
- cardiac remodelling
- interventional cardiology
- non-coronary intervention
- percutaneous valve therapy
- heart failure
- systolic heart failure
- diastolic dysfunction
- myocardial disease
- cardiomyopathy restrictive
Statistics from Altmetric.com
- Cardiac amyloidosis
- cardiovascular MRI
- cardiomyopathy
- valvular disease
- cardiac function
- cardiac remodelling
- interventional cardiology
- non-coronary intervention
- percutaneous valve therapy
- heart failure
- systolic heart failure
- diastolic dysfunction
- myocardial disease
- cardiomyopathy restrictive
Introduction
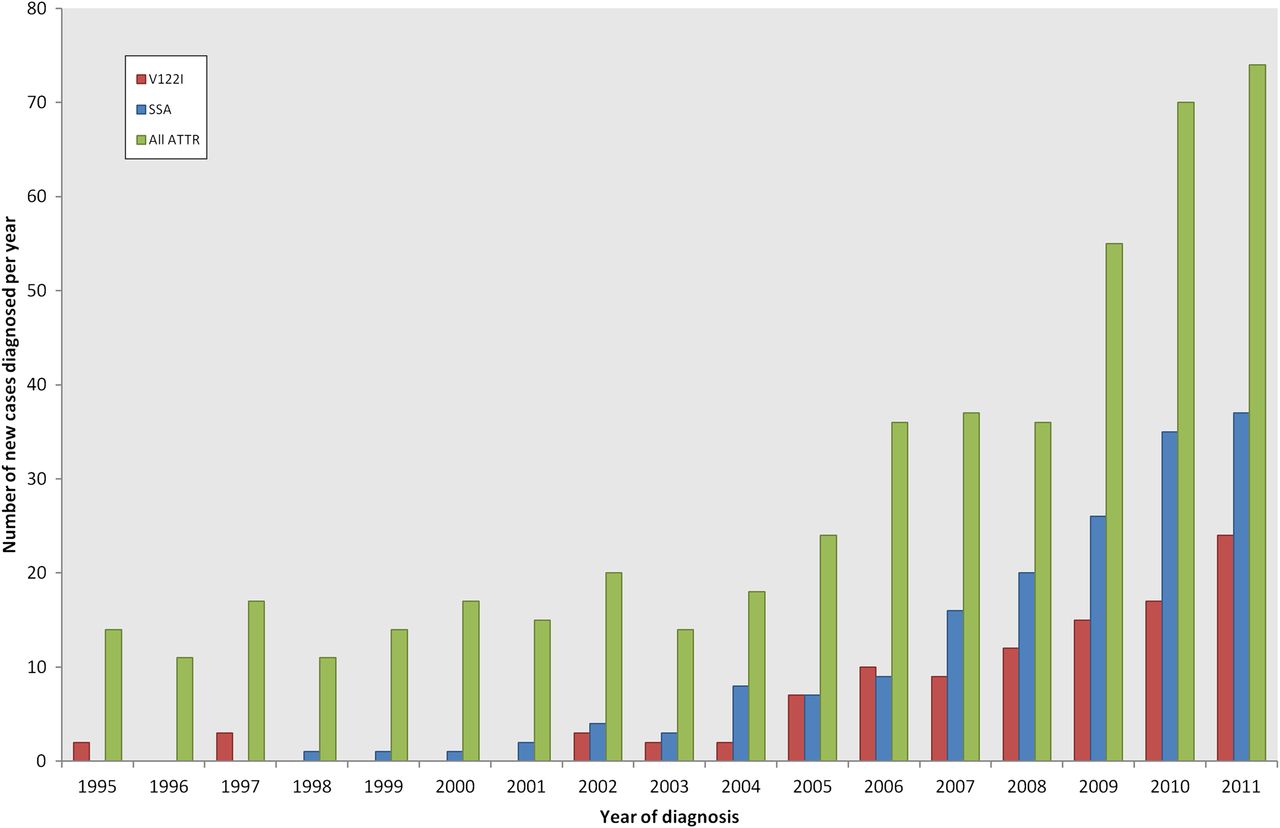
Amyloidosis is caused by the extracellular deposition in organs and tissues of proteins in a pathologic insoluble fibrillar form.1 Most of the literature relating to cardiac amyloidosis is based on the findings in patients with amyloid light-chain (AL) amyloidosis, in which the amyloid fibrils are derived from monoclonal immunoglobulin light chains. The proportion of patients with systemic AL amyloidosis presenting to the UK National Amyloidosis Centre has remained steady over a 25-year period,2 whereas we have experienced a significant increase in referrals of new patients with transthyretin-related amyloidosis during the past few years (figure 1). Transthyretin (TTR) is a 127 amino acid plasma protein that acts as a transporter protein for thyroxine and retinol-binding protein, comprising a tetramer of four identical subunits.3 TTR is predominantly synthesised in the liver but is also produced in the retina and choroid plexus.1 The TTR gene is located on chromosome 18, and about 100 amyloidogenic mutations have been described in the literature.4 TTR gene mutations were first reported in cases of Familial Amyloid Polyneuropathy (FAP); however, cardiac involvement has been described in over half of the reported variants.4 Whereas amyloidosis due to wild-type (non-mutant) transthyretin amyloid (ATTR) is a sporadic disease among older people, hereditary ATTR amyloidosis presents from the third decade onwards and is inherited in an autosomal dominant fashion, with most patients being heterozygous for normal and variant TTR.5 TTR gene mutations, which mostly result in single amino acid substitutions, are thought to promote the destabilisation of the tetrameric protein to form dissociated TTR monomers that are able to self-assemble into amyloid fibrils.6 TTR variants are usually identified in clinical practice indirectly through genetic sequencing, but the variant and wild-type proteins can be demonstrated by isoelectric focusing of serum.7

A histogram demonstrating the increasing incidence of amyloidosis of transthyretin type (ATTR) presenting to the UK National Amyloidosis Centre. Amyloid light-chain (AL) amyloidosis cases have remained steady at ∼55% of all referrals (including AL, ATTR, AA (secondary to chronic inflammatory conditions) and rarer types of amyloidosis). RED—V122I—Hereditary ATTR amyloidosis associated with variant V122I; BLUE-Senile systemic amyloidosis (SSA) associated with wild-type ATTR; GREEN—All ATTR, including SSA, V122I and all other hereditary ATTR types.
Clinical findings
Progressive breathlessness is the most common presenting complaint. Clinical signs in all forms of cardiac amyloid are predominated by right sided heart failure including lower extremity oedema, elevated jugular venous pressure, hepatomegaly and ascites. In addition, the patient may describe anginal chest pain due to diffuse microvascular amyloid deposition. Palpitations associated with paroxysmal atrial arrhythmias are common. Presentation may be with cerebrovascular events secondary to intracardiac thrombus. Syncope and light-headedness may result from hypotension due to poor cardiac reserve compounded by arrhythmia and, in the case of patients with TTR mutations causing the syndrome of familial amyloidotic polyneuropathy (FAP), autonomic neuropathy. The other typical extra-cardiac features of FAP are progressive sensorimotor neuropathy and eye involvement, typically vitreous humour opacities.
Prognosis
The overall survival of ATTR amyloidosis is significantly better than AL type with cardiac involvement. Cardiac AL amyloidosis has a high mortality with a prognosis of 6–12 months from diagnosis.8 By contrast, the prognosis of cardiac ATTR amyloidosis is typically 3–5 years from diagnosis, with one study by Rapezzi et al reporting 98% 2-year survival for hereditary ATTR and 100% 2-year survival for wild-type ATTR amyloidosis.9 The reason for the difference in survival is not clear. One theory is that the light chain-derived amyloid fibrils are toxic, resulting in myocyte dysfunction or loss and impaired cardiac function due to infiltration.10 Alternatively, faster rate of accumulation in AL amyloid might be a factor, either possibility being consistent with the development of heart failure in the setting of apparently less marked infiltration in AL amyloid.
Subtypes
Cardiac and neurological involvement in ATTR amyloidosis varies according to the mutation. Majority of the 80+ amyloidogenic mutations described in the literature present with FAP,4 in which cardiac involvement is often present, with the exception of the TTR V30M variant, the most common variant worldwide. By contrast, cardiac amyloidosis is the predominant clinical feature in senile systemic amyloidosis (SSA) and patients with the isoleucine for valine variant at position 122 (V122I). The penetrance of FAP associated variants is generally variable, while that of ATTR V122I has not been established. We review here the most common subtypes of cardiac ATTR amyloidosis presenting to the UK National Amyloidosis Centre.
Senile systemic amyloidosis (wild-type TTR)
Wild-type TTR is the amyloid fibril precursor protein in SSA.11 Wild-type ATTR amyloid deposits can be scattered widely in the body without causing symptoms, but the majority of patients diagnosed to have SSA present with a slowly progressive infiltrative cardiomyopathy.12 ,13 It is exceptional for the diagnosis to be made in individuals below 60 years of age, with typical onset beyond the age of 70 years, and there is a strong male predominance.14 ,15 In contrast to the infrequency with which wild-type ATTR amyloidosis is diagnosed clinically, autopsy studies report a 25% prevalence of senile cardiac amyloid deposits in individuals aged 80 years or over.16 ,17 This raises the possibility that the disorder often exists unrecognised in older individuals, a concept supported by increased awareness of diastolic heart failure with normal ejection fraction in this age group. The promise of specific new therapies for ATTR amyloidosis on the near horizon (see below) will serve to further raise the index of suspicion of this disorder in the older population. An important pointer to the diagnosis is carpal tunnel syndrome, which is the only reported extra-cardiac manifestation of SSA. Up to 34% of tissue samples obtained during decompression of carpal tunnel syndrome in the older population demonstrate wild-type TTR deposition, encouraging histological study in all cases.18 A history of carpal tunnel syndrome often predates SSA by some years, and should be considered a red flag in older patients presenting with heart failure and left ventricular wall thickening.19
Hereditary cardiac amyloidosis (variant TTR)
The two most commonly encountered genetic variants in our practice are ATTR V122I and ATTR T60A. Variant ATTR V122I (also known as Val122Ile or Isoleucine 122) results from the substitution of isoleucine for valine at position 122 and is associated with a phenotype that cannot be distinguished from SSA. Variant ATTR T60A (also known as Thr60Ala or Alanine 60), resulting from the substitution of alanine for threonine, is associated with FAP and major cardiac involvement in virtually every case. We discuss the phenotypes in more detail below.
ATTR V122I
The TTR V122I variant was first identified in 1988 as amyloid fibrils from a 68-year old African-American man who died of cardiac amyloidosis.20 He had no known family history of amyloidosis. DNA analysis revealed that he was homozygous for the mutation encoding the ATTR variant V122I.21 In a molecular epidemiological analysis, Jacobson et al found 66 TTR V122I alleles in DNA samples from 65 of 1688 black Americans.22 The calculated allele frequency was similar for all geographical areas in the USA, equating to 3.9% of African-Americans, that is about 1.3 million US black patients possessing the V122I allele. A subsequent study reviewing autopsy specimens supported the association with cardiac amyloidosis.23 The prevalence of variant ATTR V122I has not been established in prospective studies. However, it is likely to be an underdiagnosed cause of heart failure in the Afro-Caribbean population, based on subgroup analysis performed in African Americans participating in the β-Blocker Evaluation in Survival Trial (BEST). Buxbaum et al found 9 of the 91 patients (10%) aged more than 60 years and presenting in New York Heart Association (NYHA) class III/IV carried the amyloidogenic allele.24 It was not possible for the authors to confirm systemic amyloidosis in BEST participants but the higher prevalence of the genetic mutation in heart failure patients in this study suggests ATTR V122I is an important risk factor in Afro-Caribbeans.
The clinical phenotype of ATTR V122I amyloidosis appears to mimic SSA, that is, isolated cardiac disease and a relatively high frequency of preceding carpal tunnel syndrome. In contrast to hereditary amyloidosis associated with most other ATTR variants, neuropathy is rarely present. There have been two recent retrospective studies. One described the clinical features in 36 patients diagnosed in an American amyloid referral centre, highlighting the dominant cardiac involvement and improved survival compared with AL amyloidosis (27 vs 5 months).25 Another study reported retrospective analysis of two databases identifying 136 patients, confirming an age-dependent (>65 years) penetrance of clinical disease as assessed by echocardiography.26
TTR alanine 60 (Thr60Ala or T60A)
The genetic mutation associated with variant TTR T60A was first described in an Irish family in 1986, and a cluster of cases were identified in the County Donegal region of North-West Ireland.27 ,28 The typical clinical picture comprises progressive neuropathy, often with predominant autonomic dysfunction causing gastrointestinal symptoms of diarrhoea and/or constipation.29 Peripheral neuropathy is less common and may be confounded by carpal tunnel syndrome in the upper limbs, a common non-cardiac association. Cardiac involvement is almost always present at initial diagnosis and predicts poor outcomes in this patient group.29 Heart failure with concentric wall thickening, carpal tunnel syndrome and autonomic neuropathy in a patient with Irish ancestry should raise suspicion of ATTR T60A amyloidosis.
Diagnostic tests
Correct identification of ATTR amyloidosis is necessary to prevent inappropriate chemotherapy for systemic AL amyloidosis. Lachmann et al investigated 350 patients with a diagnosis of apparent AL amyloidosis and found an alternative genetic cause in 9.7%, including about 5% patients with hereditary ATTR amyloidosis.30 The presence of a monoclonal gammopathy had often been misleading in this series, suggesting AL amyloidosis, but it is important to appreciate that incidental monoclonal gammopathy of undetermined significance (MGUS) occurs in over 5% of persons aged over 70 years.31 The reported increased risk of MGUS in Afro-Caribbean patients may also exacerbate the diagnostic difficulties,32 given the prevalence of the amyloidogenic gene mutation causing variant ATTR V122I in this cohort. It is therefore imperative that patients are thoroughly investigated to both rule out AL amyloidosis and to positively diagnose ATTR type.
Electrocardiogram (ECG)
To date there have been very few studies specifically reviewing ECG data in patients with ATTR. Low voltage complexes are present in up to 71% of patients with AL amyloidosis.8 ,33 Conversely, low voltage complexes were less prevalent in ATTR T60A (16%) and SSA (29%) series to date.15 ,29 A retrospective analysis of ATTR V122I patients failed to demonstrate low voltages in any ECG,26 which may be explained by the high prevalence of LVH criteria on ECG at baseline in Afro-Caribbean patients. We have recently reported the ECG findings in 64 patients with ATTR V122I amyloidosis.34 The presenting ECG demonstrated left ventricular hypertrophy (LVH) criteria in 25% of all ATTR V122I patients, with a negative correlation between ECG voltage and duration of symptoms. Intraventricular conduction delay, atrioventricular block and atrial arrhythmias are reported in all cohorts of ATTR amyloidosis.9 ,15 ,29 ,34 First degree heart block is common in ATTR V122I (56%) and identifies patients at high risk for subsequent pacing requirement.34 Intraventricular conduction delay and atrioventricular block are less common in AL amyloidosis.8
Reduced heart rate variability on 24 h Holter monitoring is associated with autonomic nerve involvement and correlates with late onset disease in ATTR V30M amyloidosis.35 Reduced heart rate variability has been shown to predict short-term mortality in AA- and AL-type amyloid36 but the prognostic implications in ATTR type are not known, and are unlikely to be clinically relevant in SSA and V122I amyloid which are not reported to cause autonomic neuropathy.
Echocardiogram
Descriptions of the echocardiographic findings in cardiac amyloidosis have largely been based on experience in AL amyloidosis. Increased myocardial echogenicity (a granular, ‘sparkling’ or ‘speckled’ appearance of the left ventricle) and interatrial septal thickening were described by Falk et al in 13 AL-amyloid and two familial cardiac amyloid patients.37 The advent of harmonic imaging has reduced the positive predictive value of sparkling or speckled myocardium in detecting cardiac amyloidosis on echocardiography. Interatrial septal wall thickening is often difficult to measure accurately and is less common than thickening of the heart valves, biatrial dilatation and pericardial effusions in cardiac amyloidosis.8 Left ventricular wall thickness has been reported to be greater in ATTR amyloidosis, typically 16–18 mm, compared with 13–15 mm in AL type.8 ,15 ,26 ,29 Systolic function, as assessed by left ventricular ejection fraction (LVEF), is often normal. A large study by Rapezzi et al reported mean LVEF 58±13% in 61 patients with hereditary ATTR amyloidosis.9 Assessment of systolic function by LVEF has its limitations. The reduction of stroke volume in the context of concentric ‘LVH’ and reduced long axis function has been proposed as the mechanism of thick-walled heart failure with normal ejection fraction.38 Longitudinal function is often severely impaired in cardiac amyloidosis and tissue Doppler imaging studies in patients with cardiac AL amyloidosis have shown reduced diastolic velocities.39 Diastolic function is abnormal in the majority of patients with ATTR amyloidosis29 but the overlap between longitudinal systolic dysfunction and diastolic dysfunction has not been formally assessed in ATTR amyloidosis.
More recently radial strain has been shown to distinguish cardiac AL amyloidosis patients from healthy controls40 and longitudinal strain and strain rate are reduced in patients with AL amyloid, even before heart failure or left ventricular dysfunction is detected.41 We have seen similar findings in our ATTR patients with a characteristic ‘bull's eye’ strain pattern derived from speckle tracking, consistent with reduced basal longitudinal strain compared with the apex (figure 2).42

Echocardiography in a patient with variant transthyretin (TTR) V122I. Note atrial fibrillation, concentric left ventricular wall thickening (22 mm) with increased echogenicity, right ventricular thickening, biatrial dilatation and pericardial effusion in 2D images. Tissue Doppler imaging (top right) demonstrates reduced longitudinal function (S wave of the lateral wall 0.04 m/s). Longitudinal strain imaging (bottom right) demonstrates reduced basal strain compared with the apex, producing the characteristic ‘bull's eye pattern’.
Cardiovascular magnetic resonance (CMR)
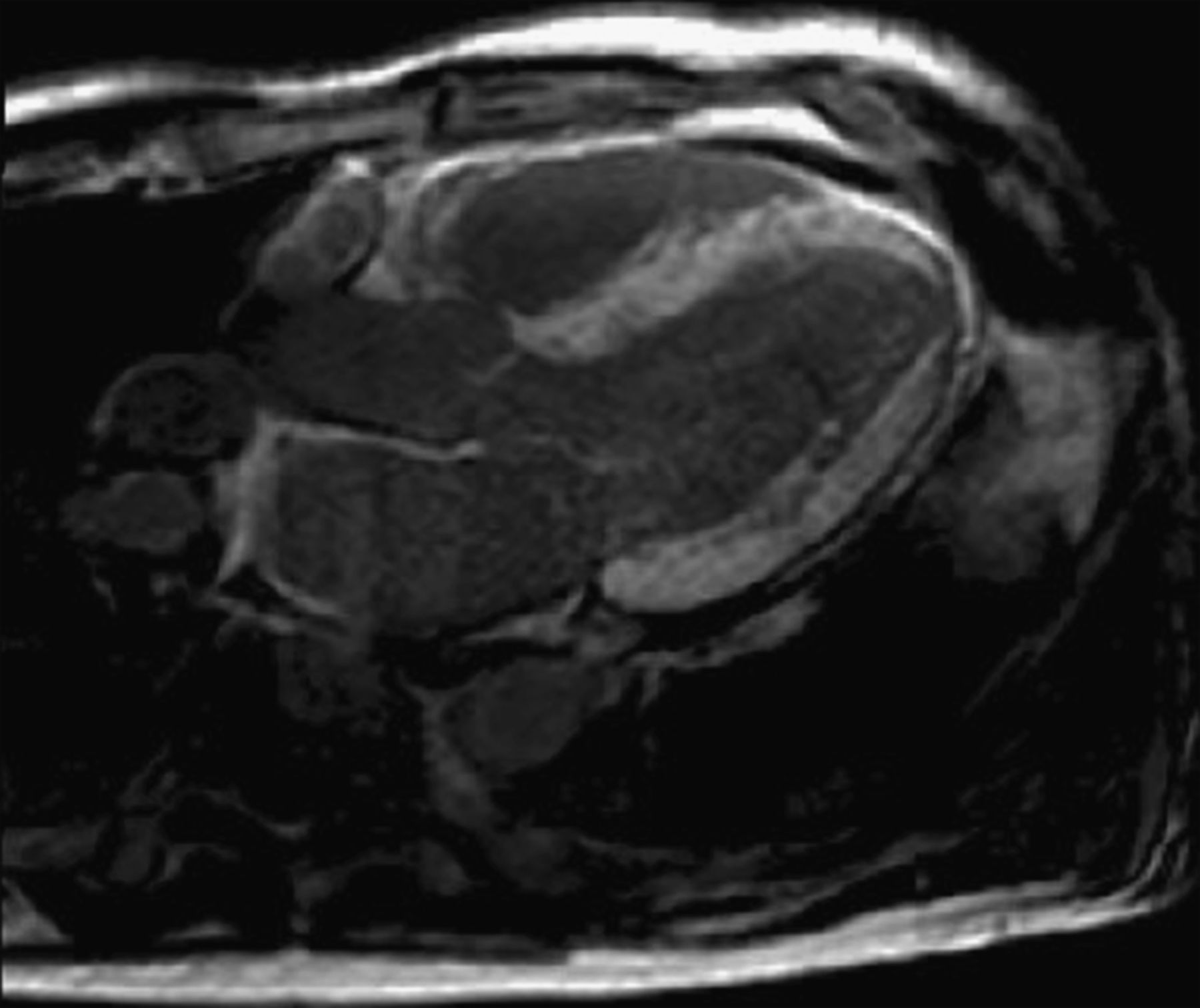
Of late, the suspicion of cardiac amyloidosis among new referrals to the UK National Amyloidosis Centre has been based on CMR, reflecting its increased availability in the investigation of heart failure and cardiomyopathy. Recent studies have reported the abnormal late gadolinium enhancement (LGE) in cardiac amyloidosis. Maceira et al studied 29 patients with amyloidosis (25 AL- and 4 TTR-amyloid) and 16 hypertensive controls.43 Following gadolinium contrast, subendocardial LGE was seen in two-thirds of amyloid patients. The authors also demonstrated shorter subendocardial relaxation time (T1) in amyloid patients and described the altered gadolinium kinetics and difficulty in nulling the myocardium. A follow-up study reported that the presence of myocardial LGE did not predict mortality.44 Other groups have suggested that the pattern of LGE is more variable, although patient numbers are relatively small.45 ,46 There have been few reported comparisons between CMR in AL and ATTR amyloidosis. We have seen a more diffuse, transmural enhancement pattern in ATTR type (figure 3) which, alongside the altered kinetics and difficulty nulling the myocardium, renders clinical studies challenging to interpret, there being no reference or ‘normal’ myocardium to delineate pathology. The use of T1 scout sequences is recommended to ensure that the appropriate inversion time is chosen for LGE sequences (See figure 4). Right ventricular LGE, present in all ATTR amyloidosis patients but absent in up to one-third of AL amyloidosis patients referred to the UK National Amyloidosis Centre, may differentiate the two amyloid types.47 Cardiovascular MRI may also have a role to play in the surveillance of patients with ATTR amyloidosis, but this is yet to be validated. While the absence of ionising radiation and ability to assess changes in left ventricular mass very accurately bode well for serial studies, repeated administration of gadolinium may be less desirable, noting that renal impairment commonly accompanies advanced heart failure. A small study by Benson et al included nine patients with biopsy-proven ATTR in whom serial CMR studies were performed at baseline, 6 months and 12 months.48 The authors report a significant increase in left ventricular mass by CMR (192 g at baseline vs 212 g at 12 months, percentage change −2 to 22, p=0.005). The increase in LV mass in this study reflects the progression of ATTR without definitive treatment of the underlying condition.
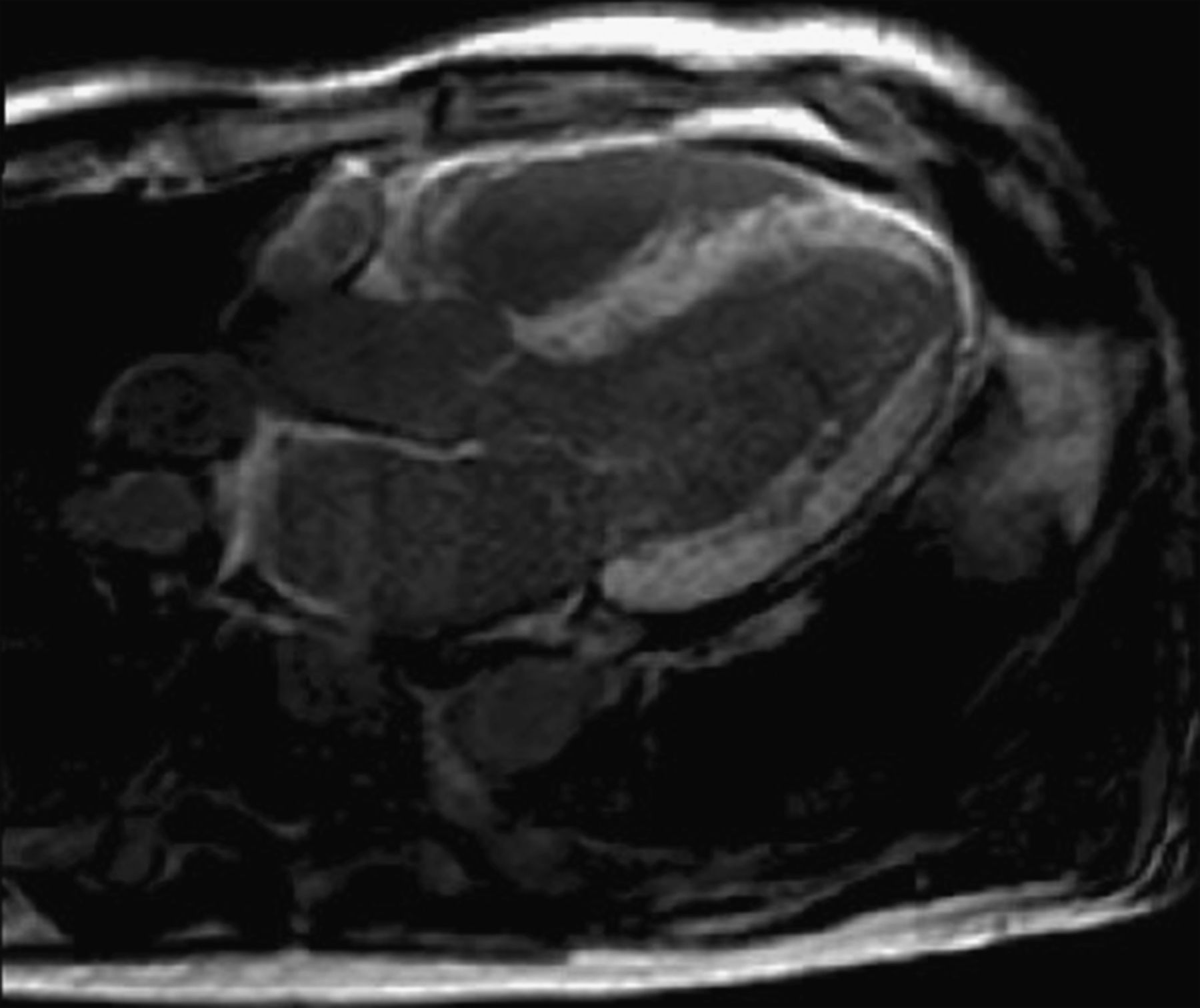
CMR—An example of diffuse transmural LGE in a patient with senile systemic amyloidosis. Serial T1-weighted scouts (Look-Locker sequences) were used to confirm that the appearance was not due to inadequate myocardial nulling. Note right ventricular and left atrial enhancement, and concentric left ventricular wall thickening.
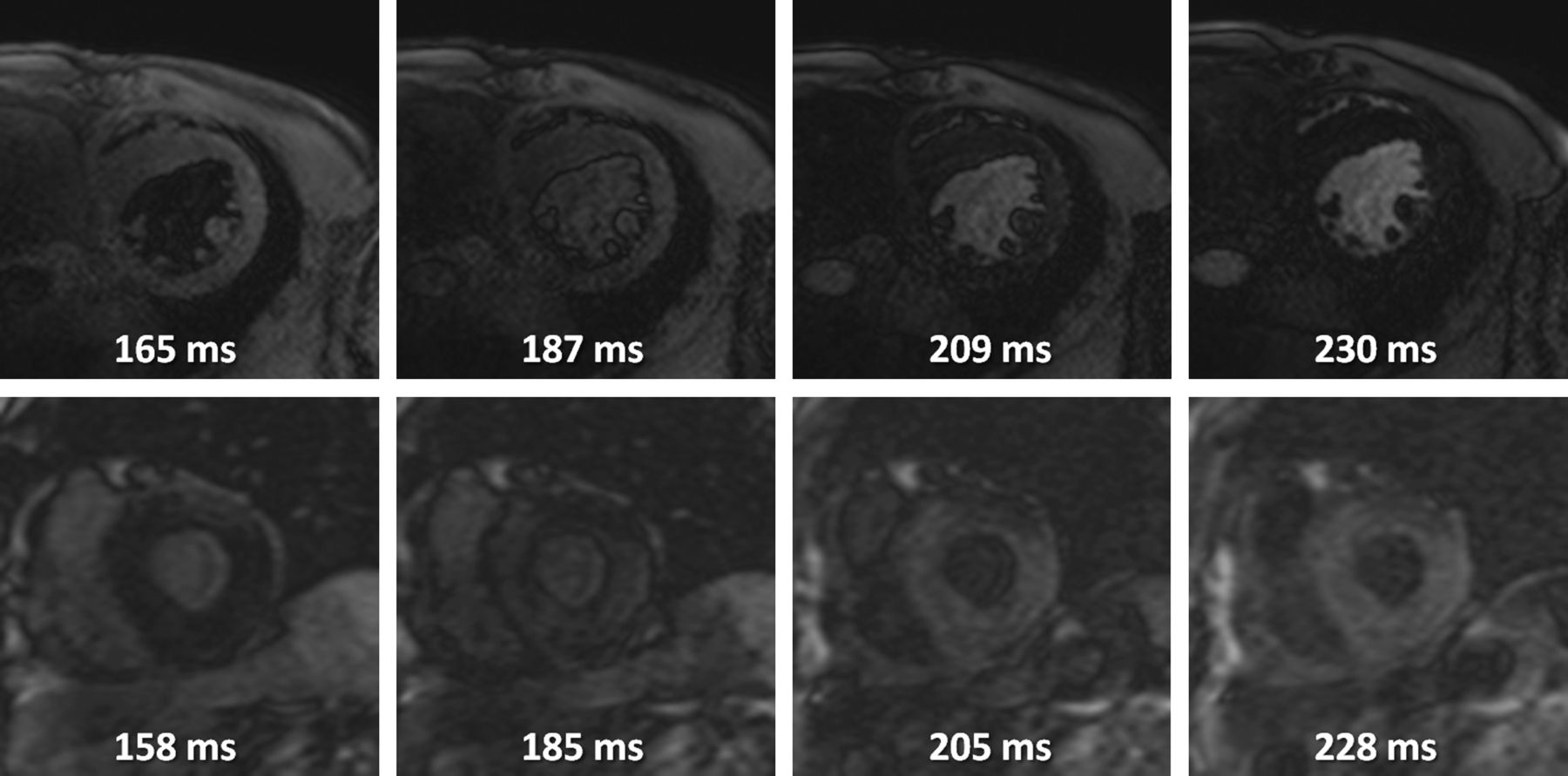
Cardiovascular magnetic resonance—Selected frames from Look-Locker inversion recovery sequences (T1 scout) demonstrating altered gadolinium kinetics in amyloidosis of transthyretin (ATTR). The top sequence is from a patient with hypertensive heart disease - the null point of the blood pool is reached before the myocardium is nulled. The bottom sequence is from a patient with senile systemic amyloidosis secondary to wild-type ATTR—the null point for the myocardium is reached before the blood pool is nulled.
Scintigraphy
Serum amyloid P component (SAP) scintigraphy
SAP is a plasma glycoprotein that binds specifically to all types of amyloid fibril. Radiolabeled SAP localises quantitatively to amyloidotic organs and SAP scintigraphy enables diagnosis and serial assessment of affected viscera.49 The pattern of organ uptake varies to some extent according to type, for example extensive bone uptake being pathognomonic for AL amyloidosis. Unfortunately SAP scintigraphy is unable to image cardiac amyloid due to blood pool background, the motility of the heart and decreased permeability of the tracer across myocardial capillary epithelia,50 and is typically negative in ATTR amyloidosis. However, SAP scintigraphy remains an important investigation to detect extra-cardiac amyloid; especially helpful among patients with AL amyloidosis in whom there is no detectable monoclonal immunoglobulin or free light chain excess to support AL type.51
99mTechnitium-labelled phosphate derivatives
Phosphonate-based nuclear medicine tracers used for bone scintigraphy have been reported to localise in cardiac amyloid, in some cases before symptoms have been present.52 ,53 The mechanism of uptake into amyloid deposits is not known, but might relate to their high calcium content.54 The 99mTechnitium labelled phosphate derivatives that have been reported to localise in amyloidosis include 99mTc-pyrophosphate (99mTc-PYP), 99mTc- methylene diphosphonate (MDP) and 99mTc-3,3-diphos- phono-1,2-propanodicarboxylic acid (DPD).55 A study by Perugini et al demonstrated more frequent uptake of 99mTc-DPD in patients with cardiac involvement of ATTR amyloid compared with AL type.52 99mTc-DPD was superior to 99mTc-MPD, which failed to localise in the hearts of all 11 patients scanned after positive 99mTc-DPD scintigraphy in this study. The most strongly positive scans all occurred in patients with ATTR amyloidosis, although subsequent studies have reported minor cardiac uptake in about one-third of patients with AL amyloidosis.53 99mTc-DPD single-photon emission CT can be used to image the distribution of the tracer throughout the heart (figure 5).
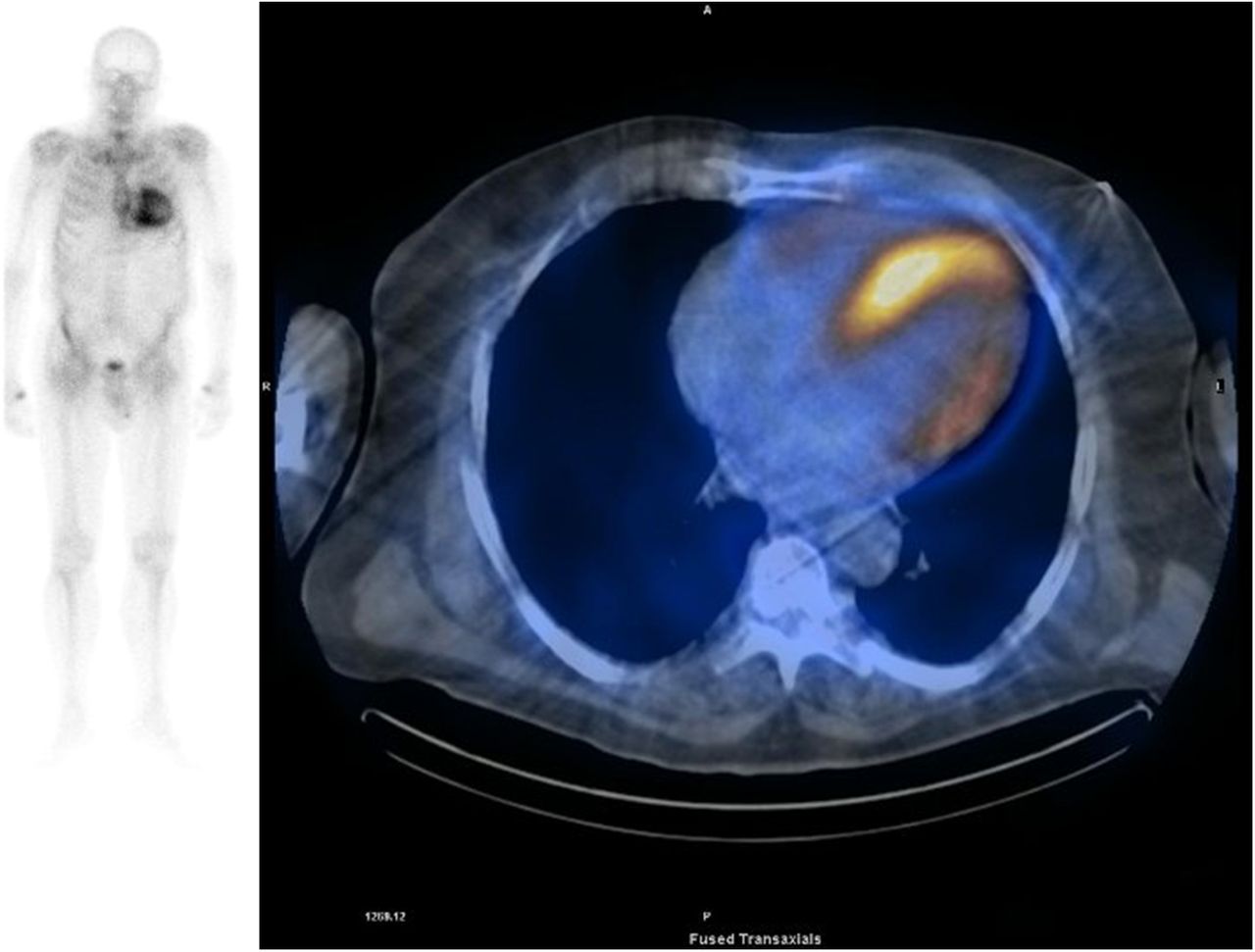
99mTc-DPD scintigraphy demonstrating myocardial uptake and attenuated bone signal on planar imaging (left panel). The right panel demonstrates fused single-photon emission CT, confirming predominant left ventricular uptake (images courtesy of David Hutt, Nuclear Medicine Technologist at the National Amyloidosis Centre).
Biopsy and need for immunohistochemistry
The diagnosis of systemic amyloidosis is confirmed through biopsy of an affected tissue or organ, and staining with Congo red to produce apple-green birefringence when viewed under crossed polarised light. The type of amyloid should then be sought with immunohistochemical staining using antibodies to a panel of amyloid fibril proteins, which usually produces definitive results in ATTR amyloidosis (figure 6), but often does not in AL type. Laser microdissection and proteomic analysis using mass spectrometry is considered the gold standard test to identify the amyloid type but is not widely available currently.56

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Endomyocardial biopsy with immunolabeled anti-transthyretin (TTR) antibody staining, demonstrating the typical ‘honeycomb’ appearance around individual myocytes (courtesy of Janet A Gilbertson, Clinical Scientist, National Amyloidosis Centre).
Rectal biopsy or flexor retinaculum biopsy at the time of carpal tunnel decompression may demonstrate amyloid and obviate the need for a target organ biopsy, for example, of the heart. However, biopsies of non-cardiac tissues tend have a lower yield in isolated cardiac ATTR amyloidosis. Abdominal fat pad aspiration is easily performed and, although older studies suggested a high false negative rate for immunotyping,14 recent studies report reasonable sensitivity using modern anti-TTR staining techniques.57
Biomarkers
TTR concentration
Serum TTR concentration is not routinely measured in clinical practice, but may provide relevant information in certain situations. A Japanese study from 1995 reported a reduction in TTR concentration following liver transplantation for FAP, and a plasma half life of variant TTR of just 2.1 days.58 A more recent study demonstrated lower serum TTR concentrations in carriers of amyloidogenic TTR mutations V30M and V122I compared with controls,59 possibly reflecting consumption of TTR into amyloid, suggesting a role in both diagnosis and surveillance of patients on new treatments.
NT-pro BNP and troponin
Serum N-Terminal pro-brain natriuretic peptide (NT-pro BNP) has been shown to be a sensitive marker of cardiac involvement and predictor of prognosis in AL amyloidosis 60 but the usefulness of this test is not well described in ATTR amyloidosis. There are some data to suggest that NT-pro BNP concentration correlates with LV wall thickness and ejection fraction in ATTR amyloidosis29 but prospective data are lacking.
Serum troponins (I or T) are detectable in ATTR amyloidosis but their role in risk stratification has not been established.61
Treatments
General management
Strict fluid balance is the mainstay of treatment in cardiac ATTR amyloidosis. Loop diuretics are generally well tolerated and patients often benefit from specialist heart failure nurse involvement, for daily weight monitoring and dose titration according to symptoms and renal function. ACE inhibitors and angiotensin receptor blockers are rarely tolerated at the maximum dose because of profound hypotension, particularly in patients with autonomic neuropathy. β-blockers should be used with caution because of the risk of decreased cardiac output, and a relatively high prevalence of conductive heart disease in patients with cardiac amyloidosis. Negative inotropic agents such as calcium-channel blockers should be avoided. Digoxin is contraindicated as it binds to amyloid fibrils and may accumulate, resulting in toxicity despite ‘normal’ levels.1 Amiodarone is well tolerated in supraventricular arrhythmias, including atrial fibrillation, but the role of Amiodarone in the treatment and prevention of ventricular arrhythmia is not known. Atrial fibrillation often occurs in the context of a rigid, enlarged left atrium, increasing thromboembolic risk. There are no absolute contraindications to anticoagulation and we recommend a low threshold for initiation in cardiac ATTR amyloidosis. Routine pacemakers for bradycardia are appropriate but the role of implantable cardioverter defibrillators and cardiac synchronisation therapy with biventricular pacemakers is not defined in this cohort.
Transplantation
Orthotopic liver transplantation (OLT) results in more than 95% reduction in the plasma concentration of variant TTR. Liver transplantation is not indicated for SSA because wild-type TTR continues to be produced by the donated organ. More than 2000 liver transplants have been performed worldwide for FAP. While this approach can halt progression of neuropathy, amyloid cardiomyopathy due to ongoing wild-type ATTR amyloid deposition appears to progress in most cases; OLT has therefore proved more beneficial in ATTR V30M associated FAP, in which cardiomyopathy seldom occurs, than in patients with other FAP causing mutations. Progression of cardiac amyloidosis following OLT has occurred in all patients under our care with the T60A variant.29 Combined or sequential liver and heart transplantation may be a solution in theory but the approach carries great risks and is rarely practicable. Despite anecdotal reports of very successful outcomes, cardiac transplantation for SSA and hereditary ATTR V122I is rarely performed in the UK, due to the advanced age of such patients (>65 years of age). Overall, transplantation has proved disappointing other than OLT in younger patients with early stage TTR V30M associated FAP, which therefore intensifies interest and need for novel drug therapies.
Disease modifying drug for ATTR amyloidosis
Several potential disease modifying treatments for ATTR amyloid are now available or poised to enter the clinic, comprising agents designed to stabilise TTR in its normal soluble conformation, and those to reduce de novo TTR production by the liver.
Diflunisal
Diflunisal is a non-steroidal anti-inflammatory analgesic which is bound by TTR in the plasma, thereby maintaining the protein in a more stable and presumably less amyloidogenic form.6 A randomised controlled trial is currently being conducted to assess its effect on progression of neuropathy in FAP.62 Diflunisal is associated with the typical NSAID-related adverse events, including gastrointestinal bleeding, renal dysfunction and worsening congestive heart failure, all of which may have devastating consequences in debilitated patients with amyloidosis. The use of diflunisal in the treatment of ATTR amyloidosis is an off-label indication and should only be prescribed in specialist amyloidosis centres with the relevant license.
Tafamadis
Tafamadis (Vyndaqel) is another small molecule that is bound by TTR in plasma and stabilises the tetrameric protein in vitro, but which has been developed specifically for FAP.63 Promising results have been obtained in a randomised double blind placebo-controlled clinical trial in 91 patients with early ATTR V30M polyneuropathy. At the primary timepoint (month 18), 45% of patients in the tafamidis group had an increase in the Neuropathy Impairment Score—Lower Limb (NIS-LL) of <2 points, compared with 30% patients in the placebo group, the differences between groups were not statistically significant (p=0.068). However, given the major unmet clinical need, tafamadis has lately been approved under exceptional circumstances by the European Medicines Agency for the treatment of TTR amyloidosis in adult patients with stage 1 symptomatic polyneuropathy to delay peripheral neurological impairment.64 It is not yet known whether Tafamidis may have a role in the treatment of cardiac amyloidosis and further studies are planned.
Novel therapies to inhibit production of TTR
ATTR amyloidosis is currently the focus of two novel approaches to selectively inhibit hepatic expression of TTR, small interfering RNA and antisense oligonucleotide therapies. The RNA silencing agent ALN-TTR01 (Alnylam Pharmaceuticals) targets wild-type and all mutant forms of TTR. A phase 1 randomised, placebo-controlled study of 31 patients (90% with variant V30M) demonstrated good tolerability and substantial reduction in circulating TTR concentration (41% relative to placebo at ∼day 7, p=0.02).65 Further studies are planned to assess the clinical effects of a much more potent agent ALN-TTR02, formulated in a second-generation lipid nanoparticle.
The competing antisense oligonucleotide approach has also entered clinical testing.66 A phase 1 dose-escalation study of the novel agent ISIS-TTRRx (ISIS Pharmaceuticals) demonstrated up to 81% reduction in TTR levels in healthy volunteers, and Phase 2 trials in patients are expected to commence in 2012.
Other novel therapies in development for amyloidosis
The compound CPHPC ((R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid) is a novel small molecule drug that cross links serum amyloid P component (SAP) in the plasma triggering its specific depletion by the liver.67 This mechanism opens a window of opportunity to target all types of amyloid with antibodies to SAP, which remains present in the amyloid deposits. The combination of CPHPC and anti-SAP-antibodies has been shown to eliminate visceral amyloid deposits in mice, and this immunotherapeutic approach is now in development in man.68
Conclusion
Cardiac amyloidosis of TTR type is emerging as an important area of clinical interest and therapeutic development. In the absence of major neurological abnormalities to raise suspicion of FAP, ATTR cardiomyopathy should be considered in any patient presenting with heart failure and marked left ventricular hypertrophy appearance on echocardiography, especially if older patients (SSA) or Afro-Caribbean (V122I) and fluid retention is in excess of that expected for the systolic function. The requirement hitherto of endomyocardial biopsy for diagnosis of cardiac ATTR amyloid, along with its non-specific appearance on echocardiography, are likely to have resulted in substantial under-diagnosis, which is now being addressed through developments in CMR and DPD scintigraphy. Cardiac ATTR amyloidosis differs from the currently far more often diagnosed AL type in its aetiology, distribution within the heart, clinical course and prognosis (table 1), and potential for treatment. It is possible that the pathogenetic mechanisms causing clinical disease may differ between the two types. Correct diagnosis, which may entail genetic testing and new proteomic analyses, is vital to ensure appropriate management, including genetic counselling of family members. Studies are in progress to optimise new CMR and scintigraphic imaging methods, determine the prevalence and significance of cardiac ATTR amyloid deposits in apparently healthy older individuals, and assess the natural history of asymptomatic carriers of amyloidogenic TTR mutations. Improved understanding of the pathophysiology of ATTR amyloidosis is ever more important with several exciting novel therapies on the near horizon. We recommend that patients with suspected amyloidosis are referred to specialist amyloid centres for further investigation, genetic screening and access to novel treatments.
Differences between cardiac amyloid light-chain (AL) amyloidosis and cardiac amyloidosis of transthyretin type (ATTR)
References
Footnotes
Funding JD is supported by a British Heart Foundation Clinical Research Training Fellowship grant no. FS/09/063/28026.
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.