Article Text

Abstract
Background The genetic basis of familial hypertrophic cardiomyopathy (HCM) is well described, but the relation between genotype and clinical phenotype is still poorly characterised.
Objective To summarise and critically review the current literature on genotype–phenotype associations in patients with HCM and to perform a meta-analysis on selected clinical features.
Data sources PubMed/Medline was searched up to January 2013. Retrieved articles were checked for additional publications.
Selection criteria Observational, cross-sectional and prospectively designed English language human studies that analysed the relationship between the presence of mutations in sarcomeric protein genes and clinical parameters.
Data extraction and analysis The pooled analysis was confined to studies reporting on cohorts of unrelated and consecutive patients in which at least two sarcomere genes were sequenced. A random effect meta-regression model was used to determine the overall prevalence of predefined clinical features: age at presentation, gender, family history of HCM, family history of sudden cardiac death (SCD), and maximum left ventricular wall thickness (MLVWT). The I2 statistic was used to estimate the proportion of total variability in the prevalence data attributable to the heterogeneity between studies.
Results Eighteen publications (corresponding to a total of 2459 patients) were selected for the pooled analysis. The presence of any sarcomere gene mutation was associated with a younger age at presentation (38.4 vs 46.0 years, p<0.0005), a family history of HCM (50.6% vs 23.1%, p<0.0005), a family history of SCD (27.0% vs 14.9%, p<0.0005) and greater MLVWT (21.0 vs 19.3 mm, p=0.03). There were no differences when the two most frequently affected genes, MYBPC3 and MYH7, were compared. A total of 53 family studies were also included in the review. These were characterised by pronounced variability and the majority of studies reporting on outcomes analysed small cross-sectional cohorts and were unsuitable for pooled analyses.
Conclusions The presence of a mutation in any sarcomere gene is associated with a number of clinical features. The heterogeneous nature of the disease and the inconsistency of study design precludes the establishment of more precise genotype–phenotype relationships. Large scale studies examining the relation between genotype, disease severity, and prognosis are required.
- Genetics
Statistics from Altmetric.com
Background
Hypertrophic cardiomyopathy (HCM) is defined by an increased left ventricular wall thickness that is unexplained by cardiac loading conditions.1 It is an important cause of sudden cardiac death (SCD), particularly in young adults, and is associated with a lifelong risk of heart failure (HF), atrial fibrillation (AF), and stroke.1 ,2 In about half of all cases, it is an autosomal dominant genetic trait caused by mutations in genes that encode cardiac sarcomeric proteins. To date, over a 1000 mutations in more than 10 genes have been reported in patients with the disease.1–11
In common with other genetic disorders, the study of associations between genotype and phenotype can shed light on the molecular mechanisms of disease and provide the basis for personalised counselling and management. Following the identification of the first sarcomeric protein gene mutations, studies of particularly large families explored the relationships between genetic variants and different subphenotypes. Specific associations were reported for individual mutations in ß-myosin heavy chain (MYH7) and troponin T (TNNT2)12–17 and some general correlations between particular genes and clinical phenotype were suggested; for example, a later onset and more benign prognosis for cardiac myosin binding protein C (MYBPC3)18–21 and mild hypertrophy and higher risk of SCD for TNNT2.22 ,23 However, subsequent work24–26 in large cohorts of mostly unrelated and consecutive probands27–32 yielded contradictory and inconsistent findings, casting doubt on the value of genetic analysis in evidence based clinical decision making.
In this paper, we summarise and critically review the current literature on genotype–phenotype associations in HCM and perform a meta-analysis on selected clinical features. The primary aim was to determine the strength of evidence for specific genotype–phenotype associations.
Methods
The methodology and presentation of the review is based on the recommendations of the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement.33
Study selection and electronic search methods
We searched PubMed/Medline up to December 2012, for the terms ‘genotype’ or ‘genetics’ combined with either ‘phenotype’, ‘age’, ‘family history’, ‘histology’, ‘wall thickness’, ‘geometry’, ‘morphology’, ‘left ventricular function’, ‘heart failure’, ‘stroke’, ‘risk factor’, ‘sudden cardiac death’, ‘syncope’, ‘abnormal blood pressure response’, ‘implantable cardioverter-defibrillator’, ‘myectomy’, ‘alcohol septal ablation’, ‘death’, ‘prognosis’ or ’outcome’, each combined either with ‘hypertrophic cardiomyopathy’ or ‘HCM’. Duplicates were eliminated. References from the retrieved original articles and reviews were searched for missing studies.
We selected observational English language human studies, with a cross-sectional or prospective design, that compared phenotypic features in patients with and without sarcomere mutations and between specific genes or individual mutations. The first selection was made by reading abstracts and the final selection by reading full articles.
In order to reduce ascertainment biases, the pooled analysis was confined to studies reporting on cohorts of unrelated and consecutive patients, where at least two sarcomere genes were sequenced. Some of these studies also included data on relatives; these data were excluded from the pooled analyses. Studies that compared specific individual mutations, data that incorporated relatives, and publications in which patients were selected on the basis of a certain phenotypic/clinical feature were excluded from the pooled analysis but were used to inform the narrative review.
Statistical analysis
Data on the following phenotypic features were extracted to a table: age; family history of HCM; histology; morphology and function evaluated by cardiac imaging (extent and pattern of hypertrophy, late gadolinium enhancement, atrial and ventricular dimensions and function, left ventricular outflow tract obstruction, mitral valve abnormalities); clinical risk factors for SCD (maximum left ventricular wall thickness ≥30 mm, abnormal exercise blood pressure response, non-sustained ventricular tachycardia, family history of SCD, syncope); interventions, outcome and prognosis (all death, cardiovascular death, SCD, non-fatal HF, AF, non-fatal stroke, implantable cardioverter-defibrillator (ICD) implantation, myectomy and alcohol septal ablation).
A random effect meta-regression model34 was used to combine the data and to obtain the pooled (overall) estimate for a set of clinical features that were most frequently reported in the final selected studies: age at presentation, gender, family history of HCM, family history of SCD, and maximum left ventricular wall thickness (MLVWT). Other clinical parameters, including the majority of the risk factors for SCD, were inconsistently reported or not rigorously defined in the great majority of the selected studies (see online supplementary table 2).
The overall prevalence was the weighted average of the prevalence across different studies. The pooled estimate is the weighted average of study specific estimates, where weights were calculated using measures of precision (inverse of the variance). Intra- and inter-study variances were used in the calculation of precision. The intra-study variance was the variance of the prevalence/proportion/mean obtained for each study. The between study variance—a parameter of the random effects meta-regression model—was estimated using method of moments. The inter-study variance was used to adjust for the heterogeneity in prevalence/proportion/mean between studies. Heterogeneity between studies was further assessed using I2 statistic, which represents the proportion of total variability in the prevalence/proportion data attributable to the heterogeneity between the studies.
Results
The initial search resulted in a total of 242 studies after removing duplicates. Twenty-two additional studies were added after reviewing the references of the retrieved articles. Reviews (n=29) and editorials (n=6) were excluded from the systematic review and publications that did not fulfil the prespecified inclusion criteria were excluded from the meta-analysis. In particular, 53 family based studies were judged to be unsuitable for pooled analyses (see online supplementary table 1) as they included small numbers of patients and were characterised by pronounced variability in the methods and scope of genetic testing, the extent of family screening, and the definition of clinical penetrance. The selection and filtering process is summarised in figure 1.

Flow chart of study selection process for the meta-analysis.
The 18 studies35–51 selected for the pooled analyses (corresponding to 13 distinct cohorts and a total of 2459 patients) are described in table 1. The pooled analysis for the percentage of individuals that are sarcomere mutation-positive (42.5%, 95% CI 35.2% to 46.7%) is shown in figure 2. Different studies were used in the analysis of individual clinical parameters, according to the completeness of the required data.
Studies selected for pooled analyses. Frequency of mutations in each sarcomere gene and sequencing methodology for each study

Pooled analysis for the proportion of individuals that are sarcomere gene mutation-positive. The overall proportion was 42.5% (95% CI 35.2% to 49.7%). The analysis shows significant heterogeneity.
Demographic characteristics and family history
Age
Younger age at presentation in carriers of sarcomere protein gene mutations is a common finding in many studies.37 ,41 ,48–51 The presence of multiple sarcomere gene mutations (double or triple heterozygosity, compound heterozygosity or homozygosity) was associated with a younger age compared to a single mutation or to the absence of mutations in a single report.41 In contrast, some other publications have reported a non-significant association between age and the presence of sarcomere mutations.42 ,43 ,46 ,47
In the pooled analysis (figure 3), the presence of a sarcomere gene mutation was associated with a younger age at presentation. The age for sarcomere-positive individuals was 38.4 years (95% CI of 28.1 to 48.7 years). The age for sarcomere-negative individuals was 46.0 years (95% CI 35.6 to 56.4 years). The overall mean difference was statistically significant (p<0.0005). There were no significant heterogeneity effects (I2=45.4% with p=0.07) between studies.

Forest plot from random effect meta-analysis showing that the presence of a sarcomere gene mutation was associated with a younger age at presentation. The age for sarcomere-positive individuals was 38.4 years (95% CI 28.1 to 48.7 years). The age for sarcomere-negative individuals was 46.0 years (95% CI 35.6 to 56.4 years) (p<0.0005). There were no significant heterogeneity effects (I2 = 45.4% with p=0.07) between studies.
Some studies reported that patients with mutations in MYH7 present earlier than MYBPC3 mutation patients,19 ,47 ,52 but others have failed to show such a difference37 ,39 ,43 ,46 ,48 ,49 or even describe a younger age at diagnosis for patients with truncating mutations in MYBPC3.37
The pooled analysis (figure 4) showed no statistically significant difference in the age at presentation between the MYBPC3 (37.6 years, 95% CI 27.1 to 48.2) and MYH7 (35.3 years, 95% CI 22.1 to 48.5) sub-cohorts (p=0.316).

Forest plot from random effect meta-analysis showing no statistically significant difference regarding the age of presentation between MYBPC3 (37.6 years, 95% CI 27.1 to 48.2 years) and MYH7 (35.3 years, 95% CI 22.1 to 48.5 years) sub-cohorts, p = 0.316. There was no significant effect of heterogeneity between studies (I2 = 50.0% with p = 0.062).
Three studies have compared age at presentation between patients harbouring MYH7 mutations and patients without mutations in this gene. In one,40 patients with an MYH7 mutation presented earlier than patients without a mutation (with or without mutations in other genes), but this was not replicated in two other studies.45 ,53 One study54 reported a younger age at presentation in TNNT2 patients than other patients with HCM. One further publication55 reported that patients with mutations in MYBPC3 were older at presentation than individuals harbouring mutations in MYH7 or TNNT2.
Gender
A higher frequency of male patients is reported in the great majority of HCM cohorts. An association between the absence of a sarcomere mutation and an even higher proportion of males is reported in two studies,46 ,50 but not in others.37 ,41–43 ,48
The pooled analysis (figure 5) showed no statistically significant difference in the proportion of males in patients with sarcomere gene mutations compared with those without (57.5% vs 61.5% respectively, p=0.422).

Forest plot from random effect meta-analysis showing the absence of a statistically significant difference between the proportion of males in sarcomere-positive (57.5%, 95% CI 53.4 to 61.6) compared with sarcomere-negative patients (61.5%, 95% CI 58.2 to 64.8, p=0.422). No significant heterogeneity effect was observed (I2 = 51.7%, p = 0.082).
There is no reported difference in the proportion of males between MYH7 and MYBPC3 mutation patients19 ,37 ,39 ,43 ,44 ,46 ,52 except for a single study that reported a higher frequency of male patients with MYBPC3 truncating mutations compared to MYH7.37 There was no difference in the prevalence of males in two studies that compared MYH7 positive to MYH7 negative patients.40 ,45
Family history
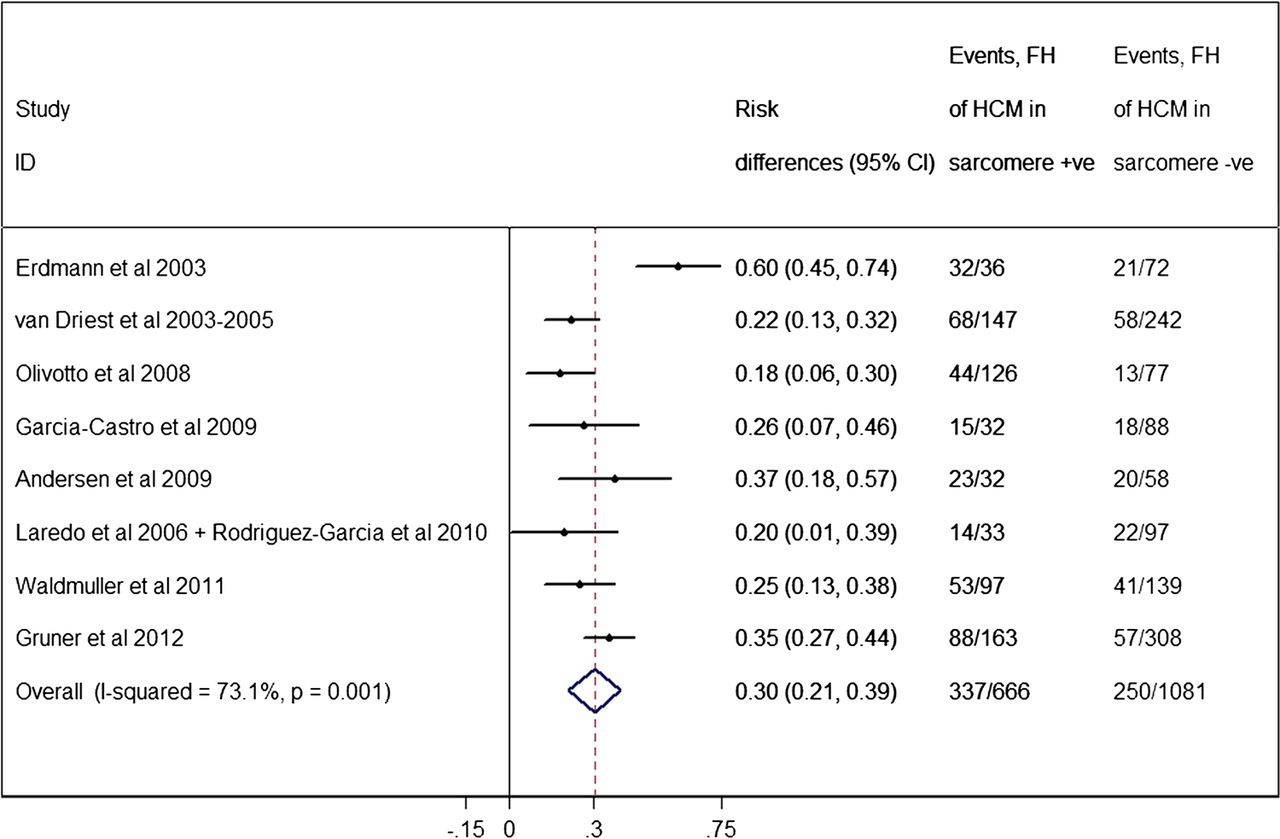
A family history of HCM has been associated with the presence of a sarcomere mutation.37 ,41 ,42 ,44 ,48 ,50 ,56 The pooled analysis (figure 6) showed a significantly higher proportion of a family history of HCM within the mutation-positive subgroup (50.6%, 95% CI of 46.8% to 54.4%) compared to mutation-negative individuals (23.1%, 95% CI of 20.6% to 25.6%, p<0.0005). There were significant heterogeneity effects (I2=73.1%, p=0.001) between studies.
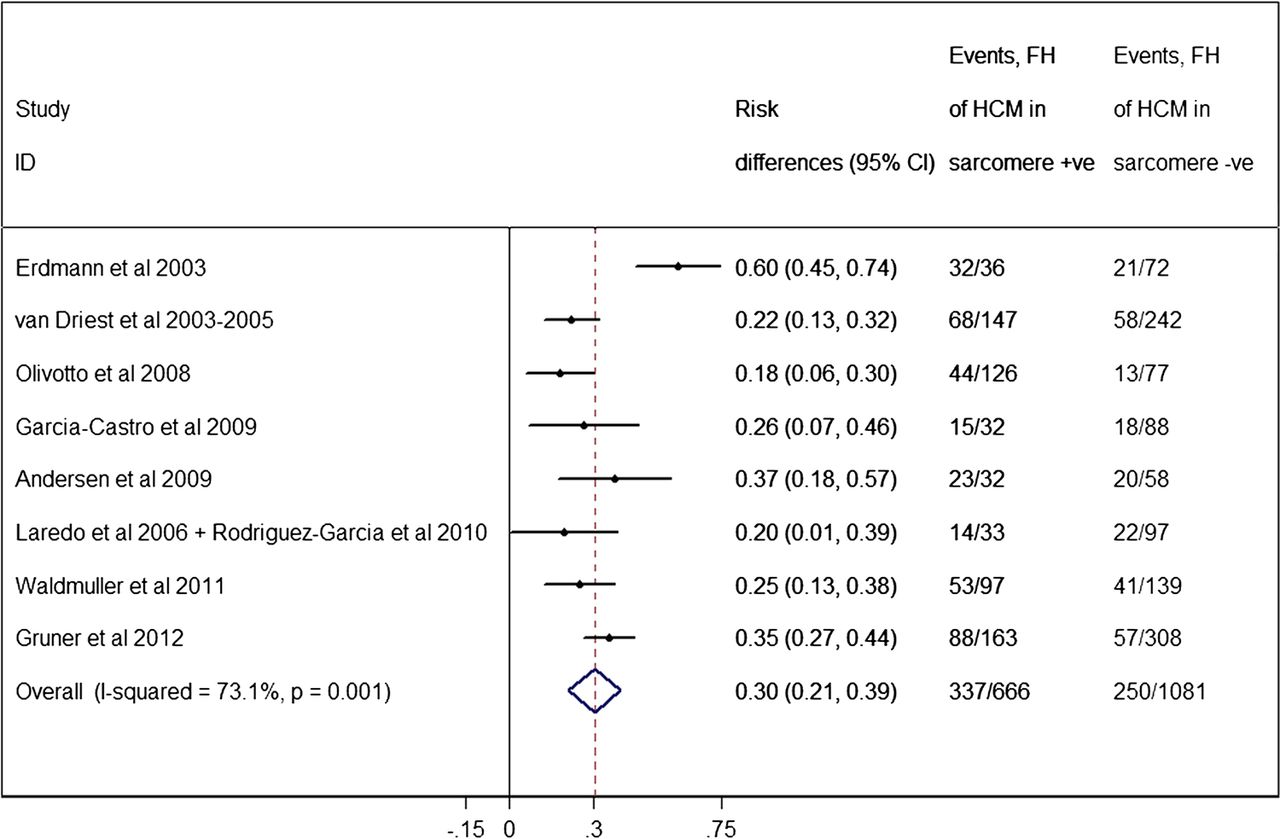
Forest plot from random effect meta-analysis showing a significantly higher proportion of a family history (FH) of hypertrophic cardiomyopathy (HCM) within the sarcomere-positive subgroup (50.6%, 95% CI 46.8% to 54.4%) compared to the sarcomere-negative individuals (23.1%, 95% CI 20.6% to 26.6%) (p<0.0005). There were significant heterogeneity effects (I2=73.1%, p=0.001) between studies.
MYH7-positive patients are more likely to present with a family history of HCM, compared with patients without mutations in this gene.40 ,43 ,45 The last study43 compared against sarcomere-negative patients.
No study has reported a difference in the proportion of a positive family history in patients harbouring MYBPC3 versus MYH7 mutations.37 ,39 ,46 ,48 The pooled analysis is shown in figure 7.

Forest plot from random effect meta-analysis showing no association between the proportion of family history (FH) of hypertrophic cardiomyopathy (HCM) in patients with MYPBC3 mutations (46.5%, 95% CI 38.6% to 54.3%), compared to the frequency of FH of HCM in patients with MYH7 (50.9%, 95% CI 41.8% to 59.9%) (p=0.284). There were no significant heterogeneity effects between studies (I2 = 54.7%, p=0.085).
In one study, patients carrying mutations in the thin myofilament genes (TNNT2, troponin I (TNNI3), actin (ACTC1), and α-tropomyosin (TPM1)) had a similar frequency of a family history of HCM compared to the rest of rest of the cohort.38
Arterial hypertension
Four studies in three different cohorts reported on the prevalence of arterial hypertension in sarcomere-positive versus sarcomere-negative individuals.44–46 ,50 Only one of those studies found a significant association,50 reporting a higher prevalence among sarcomere-negative individuals. The pooled analysis showed an overall proportion of hypertensive patients among sarcomere-positive individuals of 22.8% (95% CI 17.3% to 28.2%), compared with a proportion of 41.7% among sarcomere-negative (95% CI 37.2 to 46.2%, p=0.004) (figure 8).
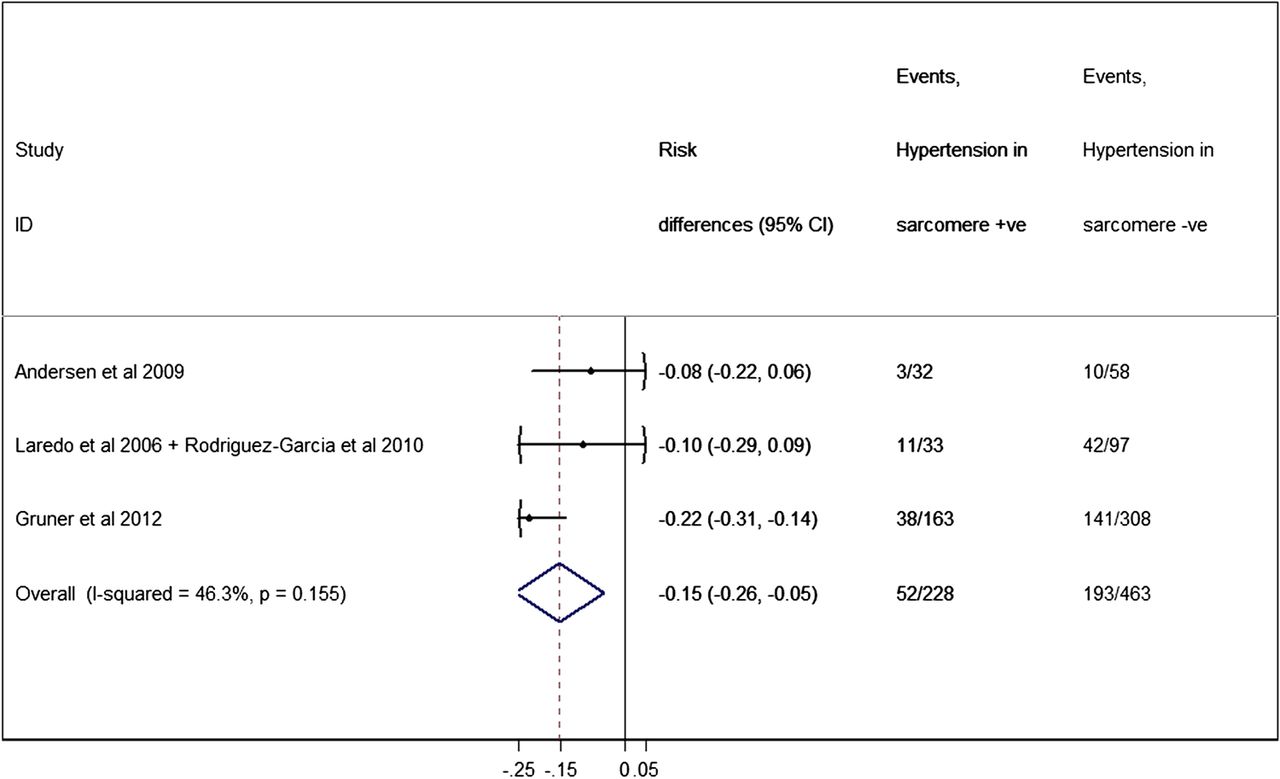
Forest plot from random effect meta-analysis showing that the proportion of patients with hypertension among sarcomere-positive individuals (22.8%, 95% CI 17.3% to 28.2%) is significantly less than the proportion among sarcomere-negative patients (41.7%, 95% CI 37.2% to 46.2%) (p=0.004). There were no significant heterogeneity effects (I2=46.3%, p=0.155) between studies.
Morphology and function
Maximum left ventricular wall thickness
The presence of any sarcomere gene mutation was associated with a greater MLVWT in several studies.41 ,46 ,49 ,50 Complex genotypes (multiple mutations in sarcomere genes) were associated with an even greater wall thickness compared to sarcomere negative patients in some but not all studies.37 ,41–43 ,48 ,51 ,57
The pooled analysis (figure 9) shows a greater MLVWT for patients carrying any sarcomere mutation (21.0 mm, 95% CI 16.9 to 25.1 mm) compared to sarcomere-negative individuals (19.3 mm, 95% CI 15.7 to 22.8 mm) (p=0.03). The heterogeneity effects between studies were not significant (I2=40.8%, p=0.133).

Forest plot from random effect meta-analysis showing a greater maximum left ventricular wall thickness (MLVWT) for patients carrying any sarcomere mutation (21.0 mm, 95% CI 16.9 to 25.1 mm) compared to sarcomere-negative individuals (19.3 mm, 95% CI 15.7 to 22.8 mm) (p=0.03). There were no significant heterogeneity effects (I2=40.8% p=0.133).
Differences in the prevalence of a maximum wall thickness ≥30 mm between sarcomere-positive and sarcomere-negative individuals were explored in two studies that found no significant associations.41 ,42
Patients carrying mutations in MYBPC3 seem to have a similar degree of hypertrophy when compared to MYH7 patients,19 ,37 ,39 ,43 ,46 ,49 with the exception of a single study48 that described an increased average LV wall thickness for MYBPC3 patients. The pooled analysis showed no difference in MLVWT between the MYBPC3 and MYH7 sub-cohorts (p=0.421) (figure 10).
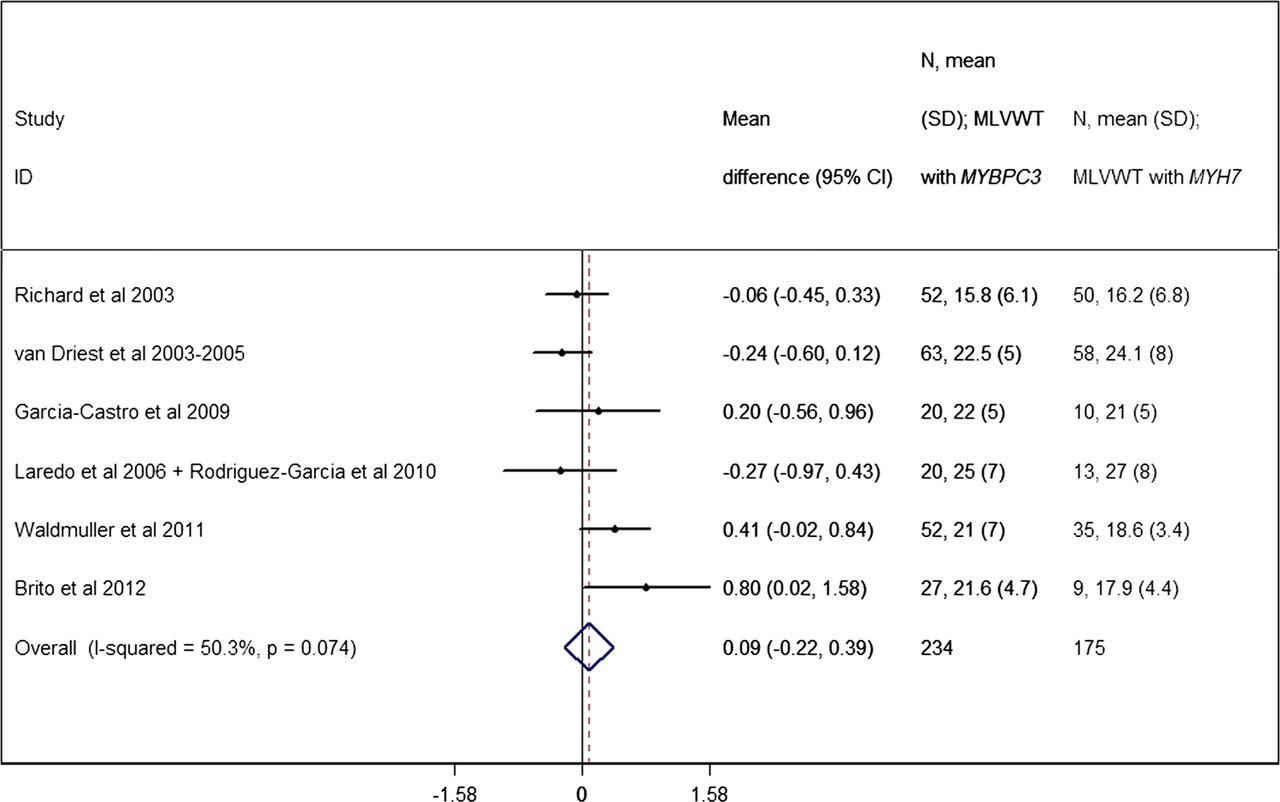
Forest plot from random effect meta-analysis showing no difference in maximum left ventricular wall thickness (MLVWT) between the MYBPC3 (21.0 mm, 95% CI 17.1 to 24.8 mm) and MYH7 (19.2 mm, 95% CI 15.6 to 22.9 mm) sub-cohorts (p=0.574). No significant effects of heterogeneity between the studies were observed (I2=50.3%, p=0.074).
In a single study, there was no difference between the LV wall thickness of patients with thin filament mutations and the rest of the cohort.38
In two studies that focus on the presence or absence of an MYH7 mutation, a higher degree of hypertrophy in MYH7 patients compared to the negative individuals is reported in one but not the other.40 ,45 Additionally, MYH7-head mutations were reported to be associated with increased wall thickness when compared to rod region mutations in another publication.52
Ventricular morphology
Few studies have analysed the distribution of LV hypertrophy. Sarcomere protein mutations are reported in two studies to be much more frequent in patients with a reverse septal curvature phenotype compared with those with sigmoidal septum,50 ,58 where this morphology was described as an independent predictor of genotype status. Another publication59 described an association between Z-disc mutations and sigmoidal septum. One study53 did not find a significant difference in the prevalence of asymmetrical hypertrophy versus other sub-phenotypes in patients harbouring MYH7 mutations.
Two studies have examined the genetic background of apical HCM. One60 reported the absence of an association between genotype and apical HCM, with some of the mutations giving rise to different hypertrophy patterns within the same family. In the second study61 there was a lower percentage of mutation-positive patients in apical HCM (13%) compared to non-apical HCM (40%).
LV dimensions and function
Most studies have not demonstrated an association between genotype and LV end-diastolic diameter, fractional shortening (FS), and ejection fraction.42 ,50 ,51 One publication53 reported a higher FS for MYH7-positive versus -negative patients and another45 reported an increased ejection fraction in patients harbouring MYH7 mutations. In a single longitudinal study,42 patients with a sarcomere protein mutation were more likely to develop LV systolic or diastolic dysfunction during follow-up.
Microvascular function and myocardial fibrosis
One study62 has reported reduced myocardial blood flow in patients carrying a sarcomeric mutation, compared with mutation-negative patients. Mutation-positive status was also associated with a higher prevalence of late gadolinium enhancement.62 Another recent publication63 described an increased level of C-terminal propeptide of type I procollagen, a biomarker of type I collagen biosynthesis, in MYH7 carriers compared to MYBPC3 carriers, without hypertrophy; this association was not present in patients with LV hypertrophy.
LV outflow tract obstruction and mitral valve abnormalities
Most studies have shown no significant association between LV outflow tract obstruction (LVOTO) and genotype status.19 ,36 ,38–46 ,49 ,52 One study50 reported an increased prevalence of LVOTO in genotype-negative patients (48% vs 37%), but it was not an independent predictor of genotype status.
The only study to report on the association between genetic background and mitral regurgitation found an increased prevalence of mitral regurgitation in MYH7 patients compared to genotype-negative patients.48
Histology
Very few studies have analysed the correlation between genotype and histologic findings. One study54 described less fibrosis and more severe myocyte disarray in patients with TNNT2 mutations; but no difference between genotype-positive and genotype-negative patients was found in a series of genotyped patients undergoing septal myectomy.64 In a study65 reporting on a TPM1 missense mutation, histopathological features were indistinguishable between the reported family and patients with other mutations. Another report66 described an association between the presence of a sarcomere mutation and the severity of cardiomyocyte hypertrophy, but not with other histologic features of HCM.
Risk factors for SCD
Syncope
Two studies described an association between the presence of MYH7 mutations and an increased prevalence of syncope in comparison to carriers of MYBPC3 mutations55 or MYH7-negative patients.53
Abnormal blood pressure response to exercise
Two studies report an association between genotype and an abnormal blood pressure response to exercise (ABPRE). In one,46 sarcomere-positive patients had a higher prevalence of ABPRE (32% in MYBPC3 and 61% in MYH7) than sarcomere-negative patients (11%). In a second,67 patients with the TNNT2 R92W mutation had a lower exercise blood pressure response compared to patients with any mutation in MYH7.
Family history of SCD
Patients carrying sarcomere mutations have a higher proportion of family history of SCD in some cohorts,42 ,47 ,50 but not others.41 ,43 ,46 ,49 Pooled analysis (figure 11) showed a significant difference in the proportion of patients with a family history of SCD between sarcomere-positive (27.0%, 95% CI 23.5% to 30.3%) and sarcomere-negative individuals (14.9%, 95% CI 12.6% to 17.2%, p<0.0005). Heterogeneity effects between studies were not significant (p=0.650).

Forest plot from random effect meta-analysis showing a significant difference for the proportion of a family history (FH) of sudden cardiac death (SCD) between sarcomere-positive (27.0%, 95% CI 23.5% to 30.5%) and sarcomere-negative individuals (14.9%, 95% CI 12.6% to 17.2%) (p<0.0005). There were no significant heterogeneity effects (I2=0%, p=0.650).
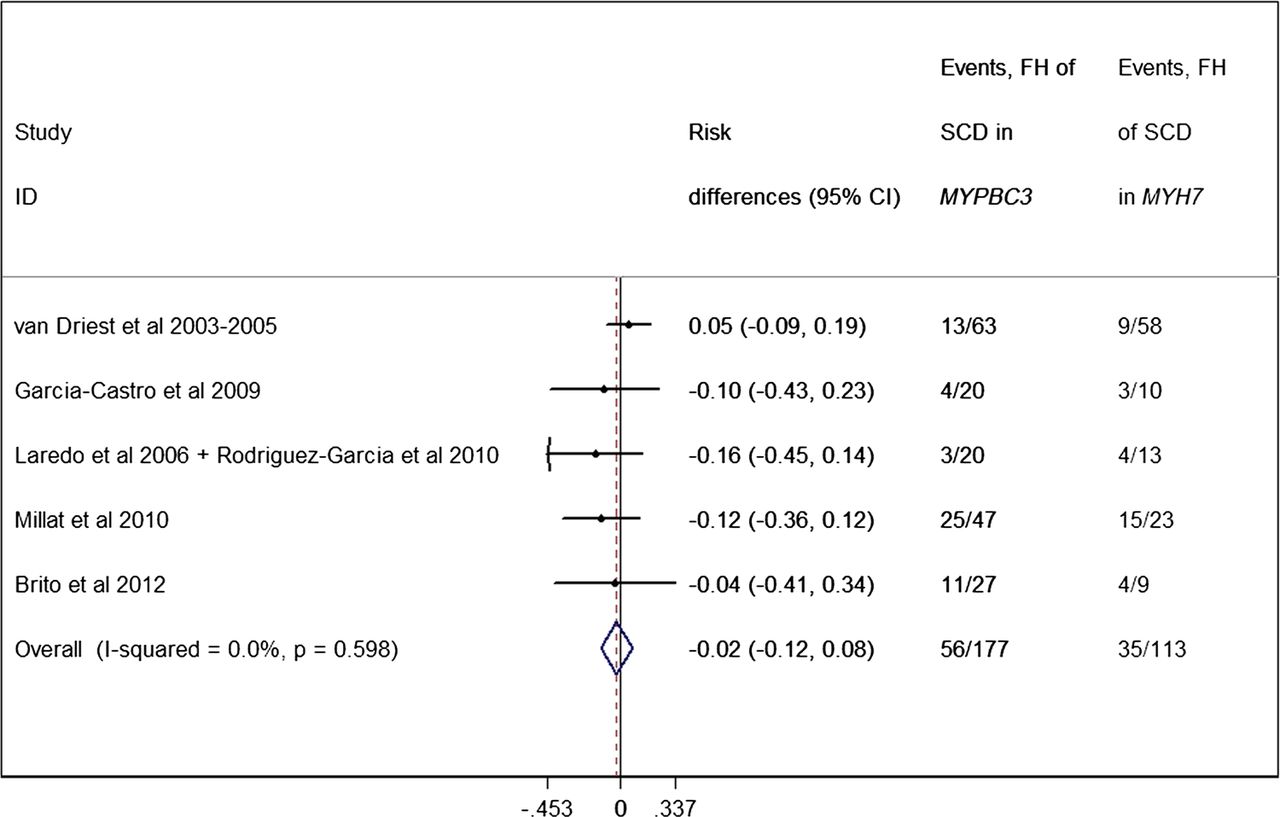
In studies comparing MYBPC3 and MYH7 mutations, only one described an increased frequency of a family history of SCD in MYH7 patients52; this was not the case in others.39 ,43 ,46 ,49 The pooled analysis (figure 12) showed no difference between MYH7 and MYBPC3 subgroups regarding family history of SCD (p=0.651).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forest plot from random effect meta-analysis showing that the proportion of a family history (FH) of sudden cardiac death (SCD) is not different between MYBPC3 (31.6%, 95% CI 24.7% to 38.5%) and MYH7 patients (31.0%, 95% CI 22.5% to 39.5%) (p=0.651). No significant heterogeneity effects were observed (I2=0%, p=0.598).
Patients harbouring mutations in MYH7 had an increased family history of SCD compared to MYH7-negative individuals in one publication,45 although the same association was not described in another.40
Interventions and prognosis
ICD implantation
Data on the frequency of ICD implantation in mutation carriers are contradictory, with some reporting an increased prevalence of implanted ICDs in sarcomere-positive patients compared to sarcomere-negative41 and others reporting no association with mutation status.42 ,43 ,49 As studies reporting ICD implants are very heterogeneous with respect to design and data presentation, a pooled analysis was not possible.
In the majority of studies comparing MYBPC3 and MYH7 mutation carriers, there are no differences in the frequency of ICD implants.39 ,43 ,48 One study37 reported an increased frequency of ICD implantation in patients with MYBPC3 truncations, and another52 reported a higher number of ICD implantations in MYH7 mutations. Finally, a study40 reported an increased implantation of ICDs in MYH7 positive patients compared to MYH7 negative individuals, which was not observed in another report.53
Septal myectomy and alcohol septal ablation
A higher frequency of sarcomere mutations is reported in one study in patients undergoing invasive septal reduction therapies alone,48 and in another study that combined treatment for LVOTO with ICD implantation.37 In contrast, three studies reported no relation between sarcomeric mutation status and septal reduction therapy.41–43
AF
The majority of studies have found no association between genetic background and the prevalence of AF.43 ,49 ,50 ,53 In one,42 chronic AF was significantly more prevalent at baseline evaluation in genotype-positive patients.
Severity of disease, prognosis, and outcome
The great majority of genotype–phenotype correlation studies reporting on outcome are family focused, analyse fewer than 100 individuals, and are cross-sectional in design (see online supplementary table 1). A pooled analysis of outcomes was not performed (see Methods).
Four studies provided longitudinal follow-up data. One was confined to individuals with MYBPC3 mutations31 (57 families and 167 genotype-positive individuals), and one to families with TNNT2 mutations32 (20 families and 92 genotype-positive individuals). Another, reporting on unrelated probands, described a poorer prognosis in sarcomere mutation-positive patients, with an increased incidence of a combined end point of cardiovascular death, evolution to New York Heart Association (NYHA) functional class III or IV HF and non-fatal stroke.42 The fourth study52 reported an increased incidence of SCD in MYH7 probands compared to MYBPC3 patients.
Some familial studies describe a particularly severe phenotype and poor prognosis in individuals with multiple mutations (homozygous or compound and double heterozygous).68–74 An increased burden of risk factors for SCD and higher likelihood of an evolution to a dilated phenotype was also reported in a series of triple mutation carriers.75
Clinical penetrance
Penetrance is very variably reported in family studies. Some describe penetrance for the whole family, while others only for adults. Some examples of general associations between genotype and penetrance are reported for MYBPC331 ,76 (late onset and higher rate of incomplete penetrance in females), MYH777 (age dependent penetrance), and TNNI3 (high rate of incomplete penetrance).30 One study19 reported later penetrance in MYBPC3 families compared to MYH7 families, but another26 described a higher than expected penetrance for a MYBPC3 mutation that generated a cryptic donor splice site.
Discussion
Given the genetic and clinical heterogeneity that characterise HCM, it is not surprising that very few genotype–phenotype relationships have been shown to be reproducible. As the great majority of mutations are private (unique to each family),11 studies lack statistical power to demonstrate correlations between individual mutations and phenotype. The variable expression of the same or similar mutations with respect to age at presentation, severity of hypertrophy, and prognosis is another important confounder.31 ,78 ,79
As this review shows, this genetic complexity has led to an exploration of genotype–phenotype effects based on the affected gene rather than individual mutations. The advantage of this approach is that it increases the sample size and power to detect associations, but as we show in this review, there are still substantial differences with respect to study design, case ascertainment and the depth of clinical phenotyping. In addition, the assignment of pathogenicity to genetic variants varies considerably and there is little consideration of other factors that might influence disease expression such as comorbidity and ethnic background.
In spite of these limitations, the meta-analysis of pooled data does show that a small number of clinical variables are consistently associated with genotype; specifically, age at presentation, MLVWT, and family history of HCM or SCD. For each of these parameters, the pooled association is significant for the comparison between sarcomere mutation-positive and mutation-negative patients, but not when comparing the two most frequently affected genes, MYBPC3 and MYH7. Patients carrying a sarcomere mutation seem to present earlier, have more severe hypertrophy, and have higher prevalence of a family history of the disease and SCD. Although data are still limited, it does seem that patients with more than one sarcomere mutation also present earlier or with a more severe phenotype41 ,51 ,57 and a higher likelihood of an evolution to a dilated phenotype,75 but this last observation requires further study.
The consistent differences observed between sarcomere-positive and sarcomere-negative patients suggest that, in spite of heterogeneity across different genes and mutations, sarcomeric protein disease might be characterised by a class effect. The corollary would be that HCM not caused by sarcomeric protein mutations has different clinical characteristics, natural history and prognosis, reflecting the large number of potential genetic and non-genetic mimics of HCM. The pooled data showing an increased prevalence of hypertension in sarcomere-negative individuals provides some circumstantial evidence for this hypothesis.
The clinical value of the associations demonstrated in this meta-analysis is probably confined at present to the general counselling of families, and in particular relatives who are considering predictive genetic testing or parents seeking reproductive advice. Specific advice on therapeutic interventions requires more robust data than exist at present, particularly in relation to the natural history of individual mutations. This will require large scale genotyping combined with new bioinformatic approaches that take into account the mutation type (eg, missense vs truncating mutations) and the biological effect of the mutation on protein structure and function, including the functional domain.80 The relatively high frequency of rare sarcomeric variation in general population databases81–84 and the influence of other co-inherited sarcomere and non-sarcomere rare variants also makes attribution of causality extremely challenging.85 Finally, more sophisticated phenotyping tools that can detect early disease manifestations (intermediate phenotypes) are required to improve understanding of genotype–phenotype correlations.
Conclusions
A pooled analysis of available genotype–phenotype data shows that the presence of a mutation in any sarcomere gene is associated with a number of clinical features. The heterogeneous nature of the disease and the inconsistency of study design precludes the establishment of more precise genotype–phenotype relationships. Large scale studies examining the relation between genotype, disease severity, and prognosis are necessary.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors All authors have contributed to the writing of this review article and are responsible for the overall content.
-
Funding LRL is supported by a grant from the Gulbenkian Doctoral Programme for Advanced Medical Education, sponsored by Fundação Calouste Gulbenkian, Fundação Champalimaud, Ministério da Saúde and Fundação para a Ciência e Tecnologia, Portugal.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.