Article Text

Abstract
Objective Improved diagnostic tools, timely closure of the shunt and a better understanding of the complexity of Eisenmenger syndrome (ES) have led to improved care and treatment in tertiary centres. These may have decreased the incidence of ES and improved survival of patients with ES, although evidence is still lacking. The aim of this study was to investigate temporal changes in incidence, prevalence and mortality in patients with ES for 35 years in the Nordic region.
Methods This was a retrospective population-based study including 714 patients with ES. Survival analysis was performed based on all-cause mortality and accounting for immortal time bias.
Results The incidence of ES decreased from 2.5/million inhabitants/year in 1977 to 0.2/million inhabitants/year in 2012. Correspondingly, prevalence decreased from 24.6 to 11.9/million inhabitants. The median survival was 38.4 years, with 20-year, 40-year and 60-year survival of 72.5%, 48.4%, and 21.3%, respectively. Complex lesions and Down syndrome were independently associated with worse survival (HR 2.2, p<0.001 and HR 1.8, p<0.001, respectively). Age at death increased from 27.7 years in the period from 1977 to 1992, to 46.3 years from July 2006 to 2012 (p<0.001).
Conclusions The incidence and prevalence of ES in the Nordic region have decreased markedly during the last decades. Furthermore, the median age at death increased throughout the study period, indicating prolonged life expectancy in the ES population. However, increasing age represents decreased incidence, rather than improved survival. Nonetheless, longevity with ES is still shorter than in the background population.
- Eisenmenger syndrome
- pulmonary arterial hypertension
- adult congenital heart disease
- epidemiology
- survival
Statistics from Altmetric.com
- Eisenmenger syndrome
- pulmonary arterial hypertension
- adult congenital heart disease
- epidemiology
- survival
Introduction
Eisenmenger syndrome (ES) is a late and almost inevitable complication to an unrestrictive, unrepaired systemic-to-pulmonary shunt characterised by pulmonary arterial hypertension (PAH) with pulmonary vascular resistance at or exceeding the systemic level with reversed shunt.1 The most visible clinical features in patients with ES are central cyanosis and clubbing, whereas severe exercise intolerance is the most obvious symptom.2
Although more common in an earlier era, when corrective surgery was not available or even possible, ES is believed to be less frequent after the evolution of diagnostic tools and surgical techniques. Thus, the prevalence of ES in tertiary centres has declined from approximately 8% of patients with congenital heart disease in the 1950s to 4% in the 2000s.1–4 However, new cases of ES still occur in patients unsuitable to repair3 5; moreover, the true prevalence as well as the incidence of ES is unknown.
Although survival rates of patients with ES are available, the majority emanate from tertiary centres and results vary and may be influenced by referral bias.6–11 Data from national registries also exist,12–14 but no population-based outcomes are available. Furthermore, recent research implies that the estimated survival has likely been overestimated for decades due to immortal time bias in statistical analyses.10
The four Nordic countries of Denmark, Sweden, Norway and Finland currently have a total population of 25.5 million. The welfare system in the countries with free access to qualified healthcare together with the civil registration number and extensive population-based registries creates a unique opportunity for a long-term follow-up of large population-based patient cohorts. The present study reports outcome data from the first, population-based cohort of unselected patients with ES for more than three decades to describe the incidence, prevalence and survival rates in these patients.
Methods
Patients with ES in the Nordic countries (Denmark, Sweden, Norway and Finland) were identified through an extensive search of the national registries. The national registries were screened for all patients registered at any hospital with pre-specified diagnose codes that could indicate ES (see online supplementary file 1) between January 1977 and December 2012 in Denmark and Finland, between January 1987 and December 2012 in Sweden, and between January 1990 and December 2012 in Norway.15 In order not to falsely exclude any patient with ES, very wide search criteria were used. Patients in whom the diagnosis of ES was confirmed by detailed review of individual medical records were included and retrospectively reviewed according to protocol.
ES was defined as the presence of a congenital pulmonary-to-systemic shunt and PAH accompanied by cyanosis. Cyanosis was defined as a mean oxygen saturation at rest of <92% and/or<87% during exercise or described clinical cyanosis.1 2 16–18 Patients with surgically corrected shunts without residual shunt but with persistent PAH were not included in the study.
The patients were divided into underlying simple and complex lesions. The simple lesions were further subdivided into pre-tricuspid (atrial septal defects) and post-tricuspid (ventricular septal defects (VSD), patent ductus arteriosus and aortopulmonary window). Complex lesions included atrio-ventricular septal defect, univentricular hearts, transposition of the great arteries and truncus arteriosus.
For evaluating the temporal change in age at death, we divided the 35-year-long period into three eras. The first era was from 1977 to the end of 1992, corresponding to the period evaluated by Corone et al.19 The second era was from January 1993 until June 2006. The third era was from July 2006, corresponding to the publication of the BREATHE-5 trial20 and the introduction of advanced therapy (AT) in ES.
Ethics
The study was registered and approved by appropriate health, ethic and data authorities in all participating countries, registered at ClinicalTrials.gov (INCT01976533). All data were anonymised when shared across borders. As this was a retrospective study evaluating existing data from medical records, individual informed consent was not required.
Statistical analysis
Results are presented as count and percentage for categorical data, and the χ2 test was used for comparing distributions. Continuous data are presented as mean±SD or median (IQR), according to distribution. The Student’s t-test was used to compare two groups, whereas the one-way analysis of variance was used to compare more than two groups. The Bonferroni correction was applied in multiple comparisons.
Prevalence and incidence were calculated based on population data from the World Bank available online (http://data.worldbank.org/indicator/SP.POP.TOTL). Incidence was calculated based on the year of first confirmed diagnosis in the medical charts. Because of the study design, incidence is only reported starting in 1977 as the incidence in earlier years may be biased by patients dying prior to 1977, thus not identified and included in this study. Prevalence is reported as the proportion of disease cases in the Nordic population at a given time. An interpolated standard line, including the 95% CI, was calculated using non-linear regression. The mean age of the study population was defined as the mean age at the end of follow-up or death.
For the survival analysis, all-cause mortality was included in the Kaplan-Meier analysis. Patients who underwent heart-lung or lung transplantation or were lost to follow-up were censored at the time of transplantation or at the time of the last contact with the national hospital system. The date of the earliest confirmation of diagnosis was used as the starting point in the survival analysis.
Overall survival analysis was performed using age as the ‘time-to-event’ variable accounting for left truncation, that is, immortal time bias. Left truncation was accounted for by specifying age at start and end of follow-up, in addition to the usual ‘status’ variable in the survival model as follows: Survfit(Surv(start_age, end_age, status)~1). Survival curves were assessed and compared with the Cox proportional hazards model according to clinical subgroups. The proportional hazard assumption was graphically verified for all covariates. In cases where the proportional hazards assumption was not fulfilled, the model was stratified for the variable. Survival curves were compared using the log-rank test. A matched control cohort from the background population was acquired from Statistics Denmark. Controls were matched 20:1 according to sex and year of birth.
The null hypothesis was rejected based on two-sided p values <0.05. All analyses were performed using R Studio, R V.3.2.2 (The R Foundation for Statistical Computing).
Results
The national registries identified 6767 patients with potential ES. Data from registries, discharge letters and medical records for all 6767 patients were screened in order to exclude patients with non-ES. Subsequently, the medical records of 2054 patients were systematically reviewed. A total of 714 patients fulfilled the ES criteria and were included in the study (figure 1). Sixty-two per cent were female, and 36% had Down syndrome. The median follow-up was 22 years (IQR: 10–33 years), corresponding to 15 911 patient-years. One patient crossed borders within the Nordic region and follow-up continued after emigration. Two patients (0.3%) emigrated out of the Nordic region and were lost to follow-up. Demographic data are presented in table 1.
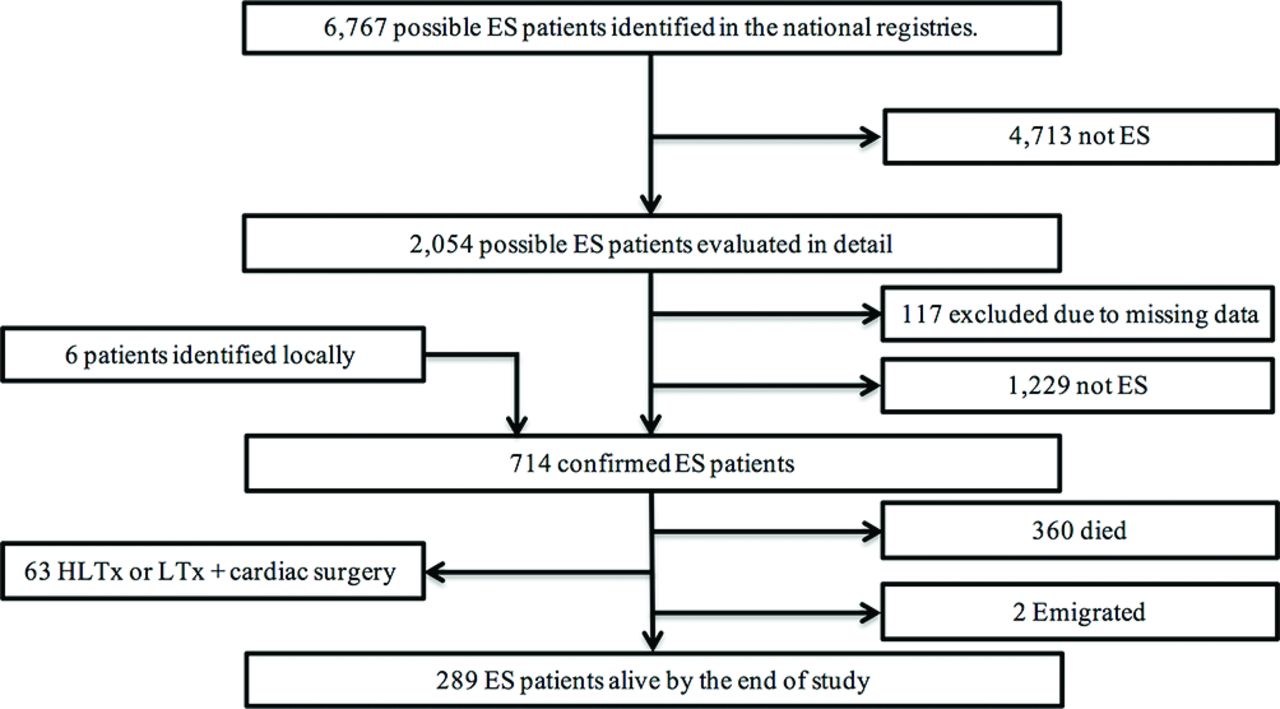
Flow chart illustrating the identification process of patients with ES in the Nordic countries from 1977 to 2012. ES, Eisenmenger syndrome; HLTx, heart-lung transplantation; LTx, single or double lung transplantation.
Demographics and diagnosis according to rank
Patients with complex lesions were significantly younger than patients with simple lesions (complex lesions: 31.0±13.6 years, simple post-tricuspid: 40.7±16.1 years and simple pre-tricuspid: 56.5±18.7 years; both comparisons p<0.001), as illustrated in table 1. Age at diagnosis was also significantly different between the shunt groups (table 1). These differences in age remained significant after adjusting for Down syndrome.
Incidence and prevalence
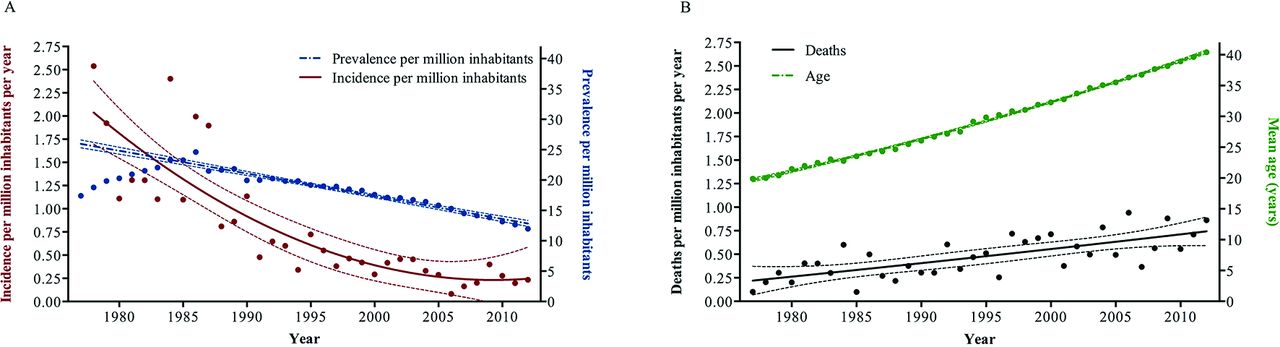
The incidence of patients with ES in the Nordic region has decreased since 1977 from 2.5/million inhabitants/year to a plateau of 0.23/million inhabitants/year over the last decade. Similarly, in the same period, the prevalence of ES has decreased from 24.6 to 12.0/million inhabitants (figure 2). The mean age of the study population increased from 19.9±11.8 years in 1977 to 40.4±15.0 years in 2012, and the rate of deaths per million inhabitants increased as well (figure 2). The absolute number of children with ES decreased throughout the study period, whereas the number of adults peaked in the mid-1990s (see online supplementary figure 1).
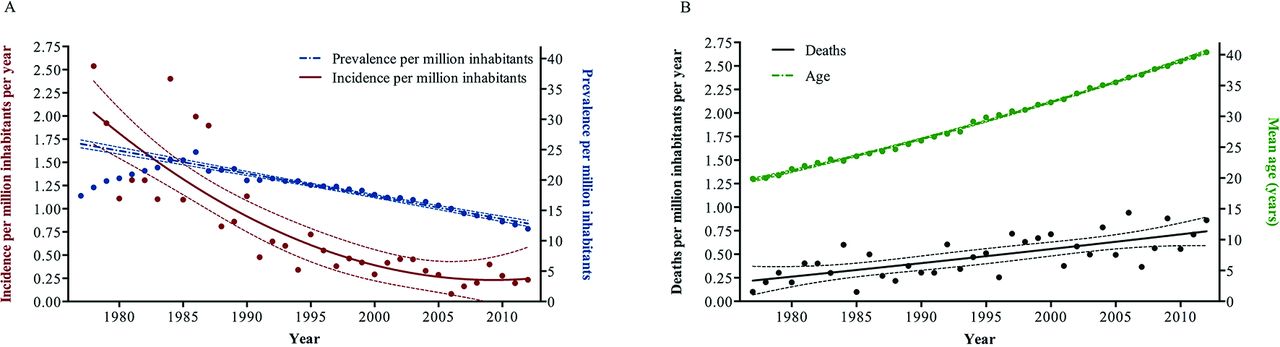
Epidemiological changes in Eisenmenger syndrome (ES) in the Nordic countries from 1977 to 2012. (A) Incidence and prevalence and (B) mean age of the ES population and the rate of death.
Survival
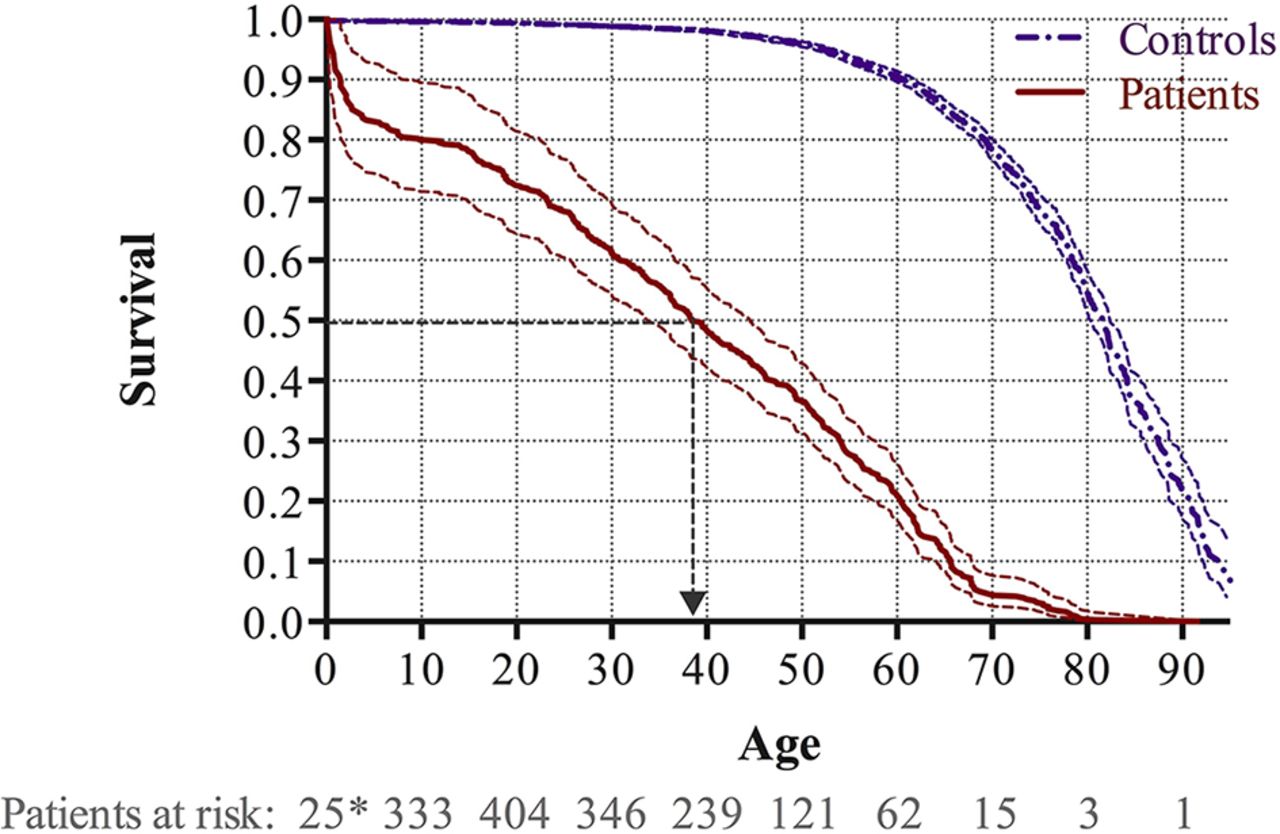
During the study period, 360 patients died, corresponding to an average annual mortality rate of 2.3%. Overall survival is illustrated in figure 3. The median survival in patients with ES was 38.4 years, which was lower than the median survival of 81.6 years for the matched background control population (p<0.001). The survival rates at the age of 20, 40 and 60 years were 72.5% (95% CI 64.5% to 81.5%), 48.4% (95% CI 42.2% to 55.4%) and 21.3% (95% CI 17.0% to 26.5%), which were significantly lower than in the matched background control population with corresponding 99.4% (95% CI 99.3% to 99.5%), 98.1% (95% CI 97.9% to 98.4%) and 90.6% (95% CI 89.7% to 91.4%), respectively. There was no significant difference between males and females (p=0.163).
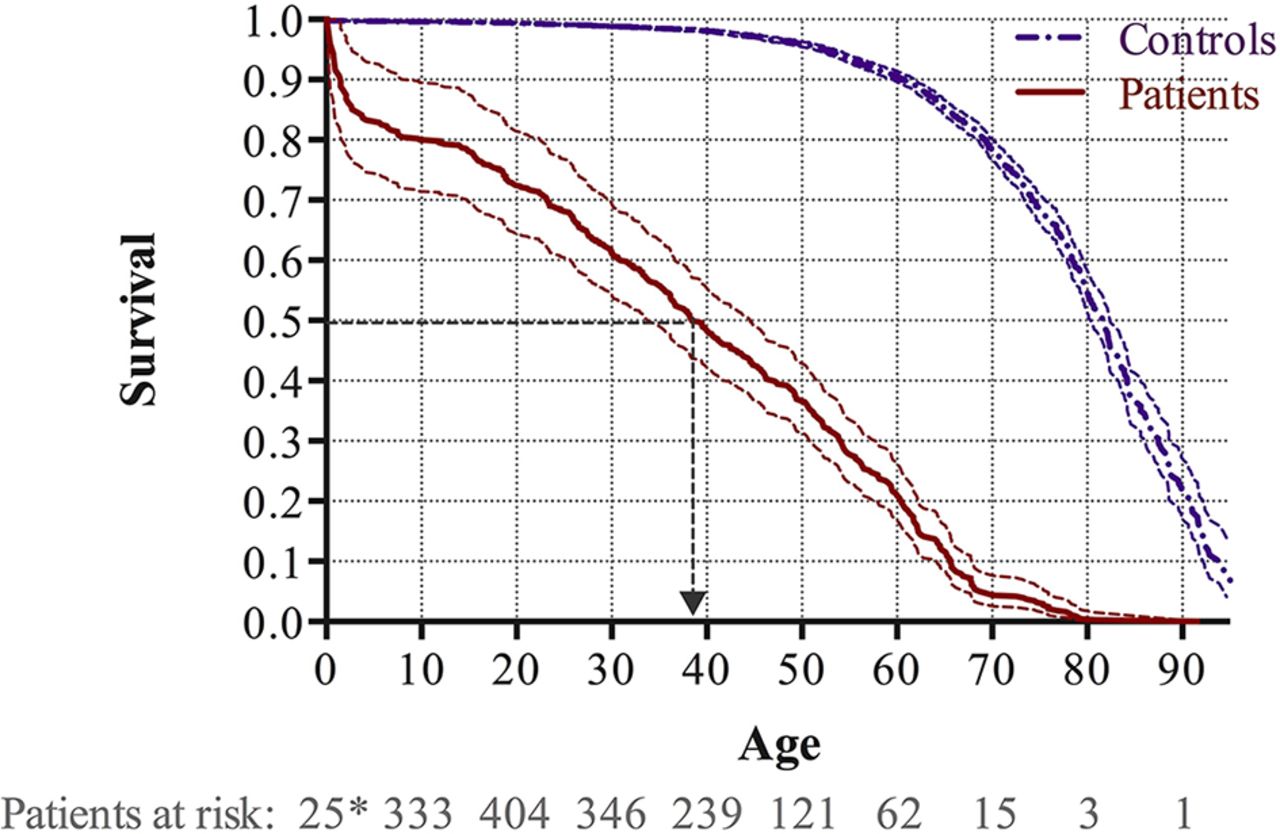
Kaplan-Meier survival curves with 95% CIs for patients with Eisenmenger syndrome and the matched background population. Survival analyses accounting for immortal time bias were employed and the number of patients at risk is provided at 1 year (*) as well as 10, 20, 30, 40, 50, 60, 70, 80 and 90 years of age.
Age at death increased throughout the study (figure 2). From 1977 to 1992 (first era), the mean age at death was 27.7±18.5 years. From 1993 to June 2006 (second era), the mean age at death increased to 38.8±19.3 years, and from July 2006 to 2012 (third era) the mean age at death further increased to 46.3±17.4 years (first vs second era, p<0.001; second vs third era, p=0.003).
Survival in simple and complex shunts
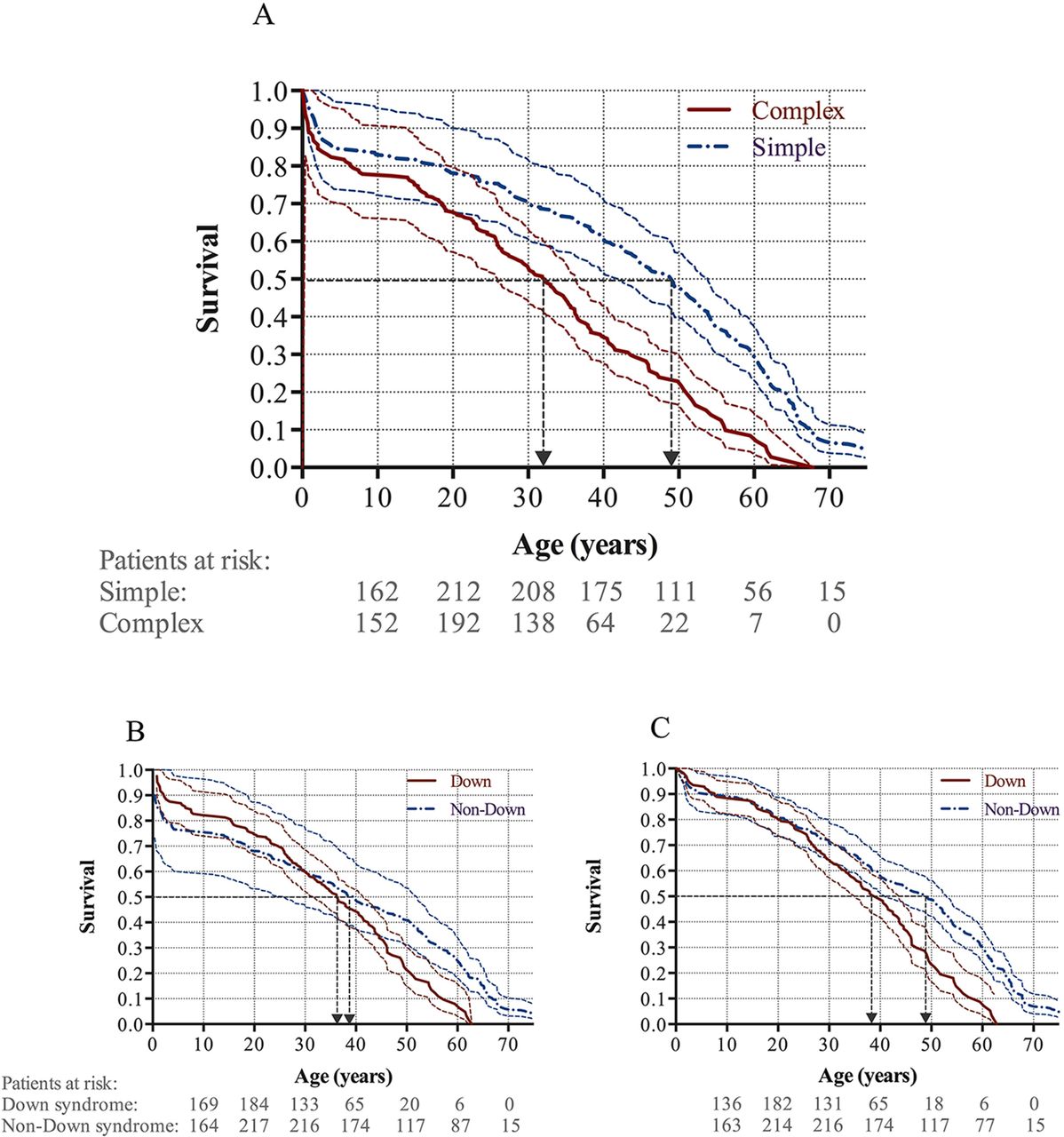
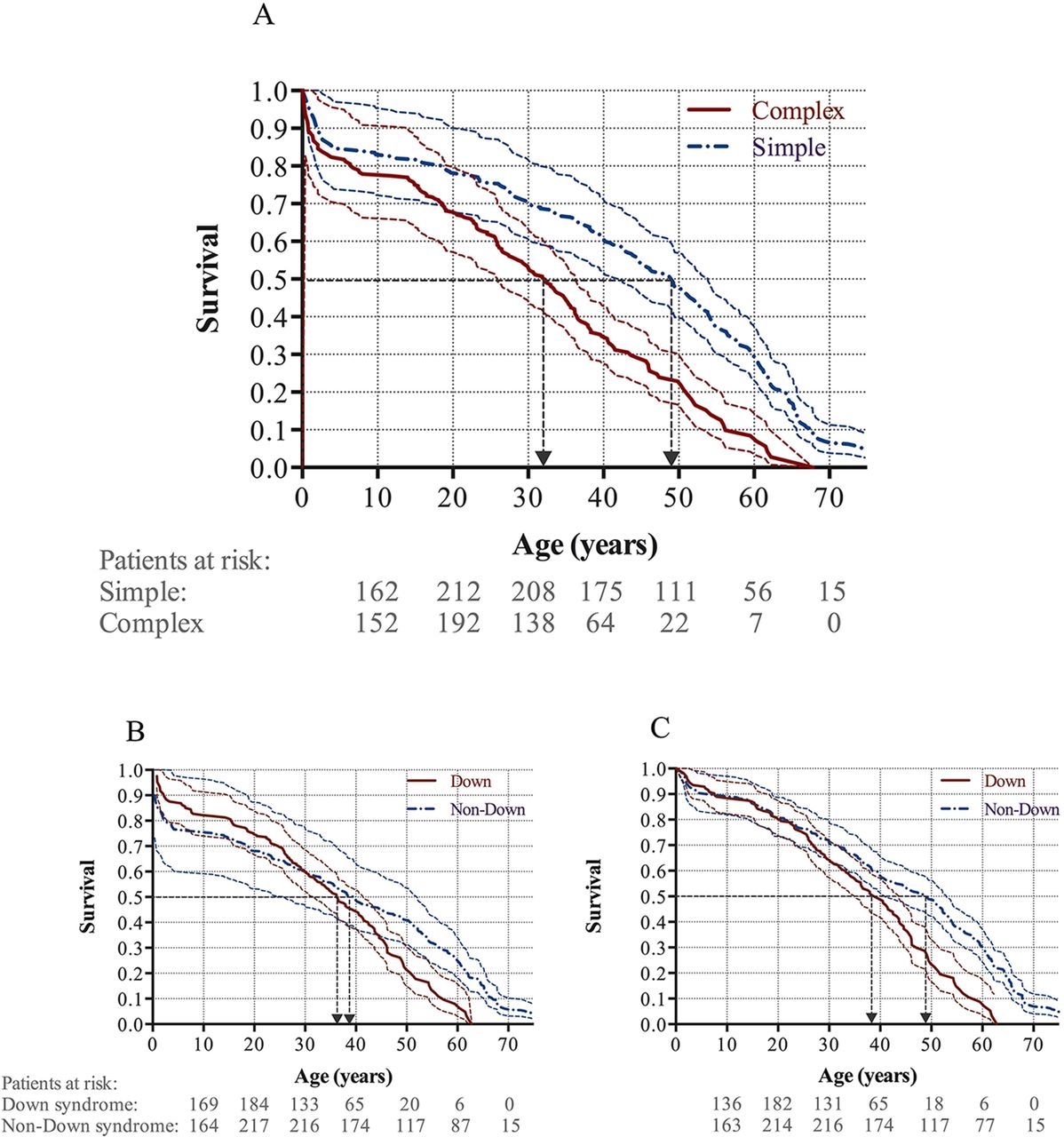
Longevity was shorter in patients with complex shunts compared with those with simple shunts, with 50% survival probability at 32.2 years vs 49.0 years, respectively (HR 2.2, 95% CI 1.7 to 2.8, p<0.001). The results were consistent after adjusting for simple pre-tricuspid shunts (p<0.001) and Down syndrome (p<0.001; figure 4A).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier survival curves with 95% CIs for (A) simple and complex shunts, (B) Down and non-Down syndrome, and (C) Down and non-Down syndrome corrected for patients dying <30 days post shunt corrective surgery.
Survival in patients with Down syndrome
Survival curves for all patients with ES with and without Down syndrome intersected at the age of 30 years; thus, the data did not satisfy the proportional hazards assumption (figure 4B). Although patients with Down syndrome appeared to have the same or even better survival than patients with ES without Down syndrome until the age of 30 years, this may have been caused by an over-representation of early perioperative deaths among patients without Down syndrome. Accordingly, when adjusting for perioperative deaths (less than 30 days after surgery) following shunt corrective surgery, there was significantly worse survival in patients with Down syndrome compared with those without (HR 1.8, 95% CI: 1.4 to 2.3, p<0.001; figure 4C).
Discussion
The main finding of this Nordic population-based study of 714 patients with ES is that both the incidence and prevalence of ES in the Nordic countries are truly decreasing. Second, it illustrates that survival in patients with ES is still poor compared with the matched background control population, despite increasing age at death during the last decades. Third, the findings of the present study confirm that patients with ES due to complex lesions still have a worse prognosis than patients with simple shunts, and finally show that ES patients with Down syndrome have a decreased longevity compared with non-Down patients.
Incidence and prevalence
The incidence and prevalence of ES have not previously been described in a large, population-based setting. The present results show that both incidence and prevalence in Nordic countries have decreased since 1977, with the incidence remaining at a constant low level around 0.19–0.25/million inhabitants/year since 2005. This finding confirms that, despite improved diagnostic and therapeutic tools, a small fraction of patients with congenital heart disease will continue to develop ES, probably due to complex shunt lesions unsuitable for repair. The prevalence of ES is continuously decreasing and currently at 12/million inhabitants, which is similar to the findings of Van de Bruaene et al in 2009 reporting a prevalence of 11/million inhabitants based on registry data from 91 patients with ES followed at tertiary centres in Belgium.21
Prevalence, incidence and duration of disease in a steady-state population are interrelated; therefore, two of these measures may be used to estimate the third. Prevalence at a certain time is, accordingly, expressed as the contemporary incidence multiplied by the expected disease duration.22 With the ageing population of patients with ES, the prevalence is likely to continue to decrease until it stabilises at a minimum level. Assuming a constant incidence rate of 0.23/million inhabitants/year and a mean duration of 38–40 years, the predicted future prevalence of ES is ~9/million inhabitants in the Nordic countries. These numbers are likely to reflect those in developed countries, but may not necessarily reflect changes in developing countries.
Survival
Several studies have evaluated the survival of patients with ES,6 7 23–25 but these estimates may be overestimated by up to 20 years due to statistical bias, as recently reported by Diller et al.10 The observed median survival rate of 38.4 years in the present study is similar to the survival rate of 34–40 years in the few available studies using the same methodology.10 14 19 The present study found a significant increase in age at death throughout the study period. However, this increase does not represent improved longevity, but rather the effect of decreasing incidence on age distribution. Thus, the population from July 2006 to July 2012 consists mainly of ‘the survivors’ of the preceding eras in addition to fewer new cases.
In comparison with other smaller cohorts of patients with ES, only two studies found a similar mean age of the study population compared with that in the present study.8 25 The remaining studies reported a considerably lower mean age of 19–35 years.6 7 9 14 23 24 26 27 This difference in demographics could indicate the ageing ES population, with the age distribution shifted to the right compared with previous cohorts,6 7 23 24 as well as to other contemporary studies.9 14 26 27 It is likely that some of the difference may represent referral bias, as other reports are all from large centres and thus may indicate that a higher proportion of younger patients with ES patients with complex lesions may be followed at tertiary centres. In support of this, simple lesions accounted for 57% of patients with ES in the present study, in accordance with a decrease in the proportion of simple defects from 65%–75% in the earliest reports6 7 23 24 to 47%–55% in the latest.14 21 26 27 This change, however, could also indicate that, in the current era, most simple shunt lesions are diagnosed and, if needed, closed before the development of pulmonary vascular disease.2 The distribution of sex, simple pre-tricuspid shunts and Down syndrome in the present study does not differ from the existing studies.6–9 14 23–27
As previously reported, we found that survival for complex lesions is almost 20 years less than for simple lesions.7 8 In contrast to our data, these earlier studies may, however, be biased by immortality, and thus have overestimated survival with up to 10 years.7 8 10 As classical Eisenmenger complex with simple VSD may be waning and future ES patients may largely suffer from complex lesions unsuitable to repair, it becomes even more important to identify specific predictors of mortality in this subgroup. Interestingly, the strongest evidence for the use of AT, which is considered one of the main improvements in the treatment of patients with ES, did not include patients with complex lesions.20
Survival in patients with Down syndrome
The survival rates for patients with ES with or without Down syndrome were, on initial evaluation, not different in the current study, as previously suggested.14 28 It was hypothesised that shunt closure was formerly not offered to children with Down syndrome29; thus, there may be an over-representation of early perioperative deaths in the non-Down syndrome patients. In contrast to the initial findings, on correction for these perioperative deaths, a statistically significant lower survival rate was found for patients with ES and Down syndrome, with a life expectancy approximately 10 years shorter than patients without Down syndrome. This finding, along with the fact that Down syndrome currently affects more than one-third of patients with ES, implies that adjusting for perioperative mortality may be needed in ES cohorts, where patients with Down syndrome may have been denied shunt closure. This has not been applied in previous survival analyses comparing patients with and without Down syndrome.14 19 28 Complex lesions, almost exclusively atrioventricular septal defects, are more frequent in patients with ES and Down syndrome compared with the non-Down syndrome population. Thus the prevalence of complex lesion may decrease slightly along with the decreasing birth rate of patients with Down syndrome in some countries due to termination of pregnancy based on foetal scan findings.30
Strengths and limitations
The major strength of this study is the large, population-based design, minimising referral bias, which may be a limitation in studies from tertiary centres. The extensive Nordic registries provide solid outcome data on all citizens, thus minimising ‘informative censoring’ and strengthening the analysis. Finally, the verification of diagnosis and subsequent data collection have been performed or supervised by the principal investigator, making data collection consistent throughout the different contributing centres.
This study has limitations due to the nature of the data. First, a retrospective design was used to collect data from medical records from several centres in the Nordic countries. Second, data from public registries were used, and misclassifications may have occurred. To minimise this possibility, all diagnoses were confirmed by reviewing the medical records before the patient entered the study. The definition of ES due to complex shunt may vary between studies available in the literature, which warrants careful review when comparing results from different centres. Third, confirmation of ES was not always possible because of incomplete data. These cases were not included in the study; moreover there was potentially limited access to data from patients dying early in the study period, which may have introduced selection bias. Therefore, exclusion of a minor number of false negative diagnoses may have occurred. Finally, this study did not account for immigration; thus, although limited in numbers, immigrants from developing countries diagnosed after arrival in the Nordic region may falsely increase the incidence and prevalence.
Conclusions
The incidence and prevalence of ES in the Nordic communities have decreased markedly since 1977, with a current incidence of 0.2–0.25/million inhabitants/year and prevalence of 8–12/million inhabitants/year; the incidence appears to have reached and stabilised at this rate. The median age at death increased from 27.7 2 years in 1997 to 46.3 years in 2012, which may indicate prolonged life expectancy in the ES population. However, increasing age represents the decreased incidence, rather than improved survival. Nevertheless, life expectancy among patients with ES is still significantly shorter than that in the matched background control population.
Key messages
What is already known on this subject?
The prevalence of Eisenmenger syndrome (ES) is decreasing in European centres specialised in adult congenital heart disease. Longevity of these patients has for several years been overestimated due to statistical immortal time bias, and recently Diller et al revealed that the survival has not improved since the 1970s.
What might this study add?
This study provides evidence of a markedly decreasing incidence and prevalence of ES in the Nordic countries since 1977, with a current incidence of 0.2–0.25/million inhabitants/year and a prevalence of 8–12/million inhabitants/year. Furthermore, age at death increased significantly during the study period, although life expectancy for these patients is still lower than that in the background population.
How might this impact on clinical practice?
These findings support the hypothesis that the number of patients with ES is decreasing in developed countries, for which data have previously been lacking, which may be of particular interest and use to cardiologists and researchers within congenital heart disease in order to plan future clinical service as well as research areas.
Acknowledgments
The following persons are acknowledged for their contribution with data acquisition and overall support of the study: Christina Christersson, Department of Cardiology, Akademiska University Hospital, Uppsala, Sweden; Aleksandra Trzebiatowska-Krzynska, Department of Cardiology, Linköping University Hospital, Sweden; Tom Omdal, Department of Cardiology, Haukeland University Hospital, Bergen, Norway; and Paula Lötjönen, Pediatric Cardiology, Hospital for Children and Adolescents, Helsinki University Central Hospital, Helsinki, Finland. We acknowledge Editage (www.editage.com) for English language editing.
References
Footnotes
Contributors CSH, ASJ and LS designed the study and are responsible for the overall content. CSH, KS, EN, BJ, TK, MD, ME, HH, MT and UT collected the data. CSH analysed data and wrote the manuscript. ASJ, KS, EN, BJ, TK, MD, ME, HH, MT, UT and LS critically reviewed the manuscript and provided scientific input.
Funding This study was financed by an educational grant from Actelion Denmark, a branch of Actelion Pharmaceuticals in Sweden.
Disclaimer Actelion Pharmaceuticals was not involved in the writing or editing of the report or analysis of the data.
Competing interests CSH received an educational grant from Actelion Pharmaceuticals. ASJ received a research grant and speaker’s fees from Actelion Pharmaceuticals. BJ received a speaker fee from Actelion Pharmaceuticals. UT received fees for lectures and being a member of advisory board from Actelion Pharmaceuticals. LS received research grant, as well as fee for lectures and being a member of advisory board from Actelion Pharmaceuticals.
Provenance and peer review Not commissioned; externally peer reviewed.