Article Text

Abstract
Cardiovascular imaging is essential to providing excellent clinical care for girls and women with Turner syndrome (TS). Congenital and acquired cardiovascular diseases are leading causes of the lifelong increased risk of premature death in TS. Non-invasive cardiovascular imaging is crucial for timely diagnosis and treatment planning, and a systematic and targeted imaging approach should combine echocardiography, cardiovascular magnetic resonance and, in select cases, cardiac CT. In recent decades, evidence has mounted for the need to perform cardiovascular imaging in all females with TS irrespective of karyotype and phenotype. This is due to the high incidence of outcome-determining lesions that often remain subclinical and occur in patterns specific to TS. This review provides an overview of state-of-the-art cardiovascular imaging practice in TS, by means of a review of the most recent literature, in the context of a recent consensus statement that has highlighted the role of cardiovascular diseases in these females.
- aortic and arterial disease
- congenital heart disease
- cardiac computer tomographic (ct) imaging
- cardiac magnetic resonance (cmr) imaging
- echocardiography
Statistics from Altmetric.com
- aortic and arterial disease
- congenital heart disease
- cardiac computer tomographic (ct) imaging
- cardiac magnetic resonance (cmr) imaging
- echocardiography
Introduction
Cardiovascular disease (CVD) contributes to 50% of the threefold increased risk of early death in girls and women with Turner syndrome (TS).1 The adverse cardiovascular outcomes result from an increased incidence of congenital heart defects (CHDs) (table 1), premature aortic disease, acquired coronary arterial disease and early stroke.1 2 The prevalence of CVD, mainly CHD, is increased in classical 45,X monosomy,3 4 and conventional cardiovascular risk factors are abundant.5 Nonetheless, CVD cannot be excluded by the presence (or absence) of particular karyotypes or phenotypes.
The most common congenital heart malformations and their prevalence in Turner syndrome
A high likelihood of CVD that is frequently subclinical means that all girls and women with TS should undergo screening for CVD. Non-invasive cardiovascular imaging has become essential for timely diagnosis of CVD in TS where distinct patterns of disease necessitate syndrome-specific imaging strategies6 7 Moreover, subsequent imaging is needed to detect emerging disease in ageing females with TS despite normal initial screening for CVD6 7 Evidence specific to TS for medical and surgical interventions in CVD is often lacking. However, as per expert consensus in the recent clinical practice guidelines, imaging has an important role in guiding surgical and medical therapies as well as recommendations related to sports, pregnancy and childbirth6 7
Our understanding of CVD in TS has shifted, and cardiovascular imaging is now an intrinsic part of the continuing care of girls and women with TS. Unfortunately, incomplete cardiovascular investigation remains common, resulting in missed or delayed CVD diagnoses.8 9 In view of the recently published guidelines, this review provides an overview of the use of contemporary cardiovascular imaging in TS6 7
Patterns of CVD
The pattern of CVD changes across the lifetime in TS.1 2 CVD is a principal determinant of the increased early morbidity and mortality. This is mainly due to left-sided obstructive and shunt lesions. Aortic dilation and dissection subsequently become important determinants of outcomes later in life, which is followed later on by emergent ischaemic heart disease and stroke.1 2 Owing to the changing CVD patterns, cardiovascular imaging strategies should depend on age and symptoms. Transthoracic echocardiography (TTE), cardiovascular magnetic resonance (CMR) and, in some instances, cardiovascular CT (CCT) are the essential imaging modalities (table 2). Ancillary modalities include transoesophageal echocardiography and invasive angiography.
Relative utility of non-invasive cardiovascular imaging in girls (<15 years) and adult women (≥15 years) with Turner syndrome in daily clinical imaging practice
Congenital heart disease
CHD is highly prevalent in TS. The encountered defects span from lesions that are symptomatic early in life to intially subclinical lesions that may impact prognosis later on in life. Other anomalies, such as those of the systemic venous return, are incidental and only gain importance when undertaking cardiothoracic surgery or other interventions.4 Non-invasive cardiovascular imaging has a well-established role in the diagnosis and surveillance of CHD. Because of a potentially grave impact of CHD and the important role of imaging, all patients with TS need screening for CHD at the time of diagnosis— irrespective of clinical signs and symptoms (table 3)6 7
A late diagnosis of TS in adolescents or adults is not uncommon,2 and the same systematic screening should apply to these females (table 3). The classical 45,X karyotype comes with a higher prevalence of CHD, but a non-exclusive association makes screening necessary irrespective of karyotype.3 Conversely, genetic testing for TS should be considered when imaging detects left-sided obstructive lesions in a female, because these may be independent markers of TS.10 The constellation of left-sided obstructive lesions may be very severe, such as hypoplastic left heart syndrome and Shone complex.8 9 The prognosis is poor in these conditions, and imaging strategies are defined by clinical presentation and surgical strategies. Consideration of genetic testing is important since TS is found in 1–7% of patients with hypoplastic left heart syndrome,11 and the additional diagnosis of TS heralds a worse prognosis.11–13
Valvular heart disease
TS has the highest prevalence (15–30%) of bicuspid aortic valve (BAV) of any syndrome.3–5 14 Aortic valve regurgitation or stenosis is more frequent in BAV,3 and it mainly has an adult onset owing to progressive dilation of the valve annulus and/or degenerative leaflet disease. Childhood onset aortic valve dysfunction is infrequent, as are other structural aortic valve anomalies such as congenital valvar stenosis, annular hypoplasia and subvalvar membrane.5 Importantly, the risk of aortic dilation and dissection is increased in girls and women with TS and BAV,15–18 and early diagnosis informs aortic surveillance strategies even with normal BAV function.
The BAV diagnosis is primarily made with TTE. Acoustic windows are generally favourable in children with TS but with physical growth the acoustic windows tend to become limited due to a barrel-shaped chest with lung obscuring the heart. In this context, TTE fails to ascertain valve morphology in 6% of adults.19 Given the availability of unrestricted imaging planes that are independent of acoustic windows, CMR reliably characterises aortic valve morphology using high temporal and spatial resolution, en face cine imaging of the aortic valve. The intercoronary cusp fusion predominates in TS (82–95%).19 20 Aortic valve function is usually followed with TTE due to good diagnostic performance, wide availability, radiation-free nature and low cost. However, CMR is a helpful adjunct due to high diagnostic precision for quantification of valvar regurgitation, direct planimetric valve area measurements, and for the important assessment of left ventricular volumes, mass and function that may be perturbed in abnormal loading conditions.
Mitral valve disease is infrequent. Isolated reports include prolapse, cleft, accessory, parachute and degenerative leaflet disease, as well as supravalvar stenosis.21 Pulmonary and tricuspid valve disease is even less common.
Aortic arch anomalies
Aortic coarctation (CoA) is common (12–20%) and 5–12% undergo childhood repair.3 4 22 23 Early diagnosis and treatment seek to prevent complications related to left ventricular cardiomyopathy, thoracic aortic dilation, systemic arterial hypertension, early atherosclerosis and intracranial haemorrhage (from berry aneurysm).24 25 Native unrepaired, residual or recurrent CoA is seen in 5–8% of adults—the majority of which are clinically silent and do not require treatment.3 4 15CoA can be considered a part of an anomalous arch phenotype in TS (figure 1). At the severe end of this syndrome-associated arch phenotype are arch interruptions and hypoplasia—either in isolation, cosegregated with BAV or as part of complex left ventricular outflow tract obstructive defects such as hypoplastic left heart syndrome and Shone complex.21 At the other end of the spectrum is the very common elongated transverse aortic arch (ETA) (47–49%). This anomaly is defined most easily from CT and CMR by the presence of an elongated transverse arch with an inferior kink at the aortic isthmus (the short segment by the ductus arteriosus).3 4 ETA should be noted when present because, similar to CoA, it is associated with a higher risk of aortic dilation and hypertension.26 The aortic dissection risk is increased with CoA, but this remains to be established for ETA.18

The spectrum of native and repaired anomalies of the transverse aortic arch in Turner syndrome (TS) ranges from incidental and benign to outcome-determining. (A) Cardiovascular magnetic resonance (CMR) in a 37-year-old woman with TS shows an elongated transverse aortic arch (kink at aortic isthmus (A, arrow)) with an incidental and benign aberrant right subclavian artery (A, arrowhead) arising from the posterior transverse arch. The 3D volume-rendered reformat (VR) of the non-ECG-gated contrast-enhanced angiography shows more conventional origins of the right common carotid (A, square), left common carotid (A, circle) and left subclavian (A, asterisk) arteries; (B) CMR in a 54-year-old woman with TS shows hypoplastic left-sided transverse aortic arch (B, arrow) with a dilated ascending aorta (B, asterisk) on the maximum intensity projection of the 3D contrast-enhanced non-ECG-gated angiography; (C) invasive angiography with a 2D aortogram in an 8-month-old girl with TS shows severe aortic coarctation (CoA) (C, arrow) in a left-sided aortic arch with an incidental ’bovine' arch (shared origin of the brachiocephalic and left common carotid arteries (C, square)). Percutaneous balloon dilation (C, asterisk) was performed with the subsequent 2D aortogram showing no significant residual coarctation (C, arrowhead); (D) CMR in a 48-year-old woman with TS shows moderate CoA (D, arrow) with mild transverse arch hypoplasia (D, arrowhead) and mild dilation of the descending aorta (D, asterisk) on the 3D VR of the contrast-enhanced non-ECG-gated angiography. The signal loss in the aortic root represents metal artefact from a mechanical aortic valve replacement as is better appreciated on the single frame from a 2D ECG-gated cine image of the aortic root (D, circle); and (E) CMR in a 56-year-old woman with TS shows an extra-anatomical jump graft (E, arrow) inserted for bypass of severe native coarctation (E, arrowhead) with a dilated descending aorta (E, asterisk) on the 3D VR of the contrast-enhanced non-ECG-gated angiography.
TTE non-invasively identifies and grades the severity of CoA according to luminal narrowing, post-stenotic dilation, focal flow acceleration and the derived pressure gradient, and any diastolic tail on abdominal aortic Doppler.25 The estimated gradient relies on the Bernoulli equation, which can be less reliable for a long segment CoA. 3D imaging exquisitely visualises the coarctation site and collateral arteries (which may falsely reduce the measured gradient) and should be performed for all new CoA diagnosis, including consideration of visualisaton of the intracranial vessels (berry aneurysms).24 CMR is often preferred over CCT because CMR is radiation-free, has good spatial resolution and allows functional imaging of the cardiac valves and chambers. However, CCT may be favoured when planning an intervention due to high spatial resolution. Treatment may be considered when the gradient exceeds 20 mm Hg (on TTE) and/or the luminal size reduction surpasses 50% (relative to the diaphragmatic aorta on CMR/CCT)—especially with hypertension, abnormal blood pressure response on exercise and/or left ventricular hypertrophy.24 25 Catheter angiography directly measures the pressure gradient and remains the gold standard, but the diagnostic accuracy of non-invasive imaging has decreased the use of this test. Ambulatory blood pressures should generally be performed on the contralateral arm.
When choosing CoA repair strategies in TS, it is prudent to assess for the common dilation and branching anomalies of the head and neck arteries (table 1, figure 1).3 4 Reported complication rates for percutaneous CoA interventions in TS vary, but no imaging finding quantifies any added risk for TS.27 28 Non-invasive imaging surveillance should continue after the repair—guided by clinical findings—to detect residual or recurrent CoA and repair site aneurysms. CMR combined with TTE are preferred for follow-up, but stents are best visualised with CCT due to metal artefact on CMR.
Aortic dissection
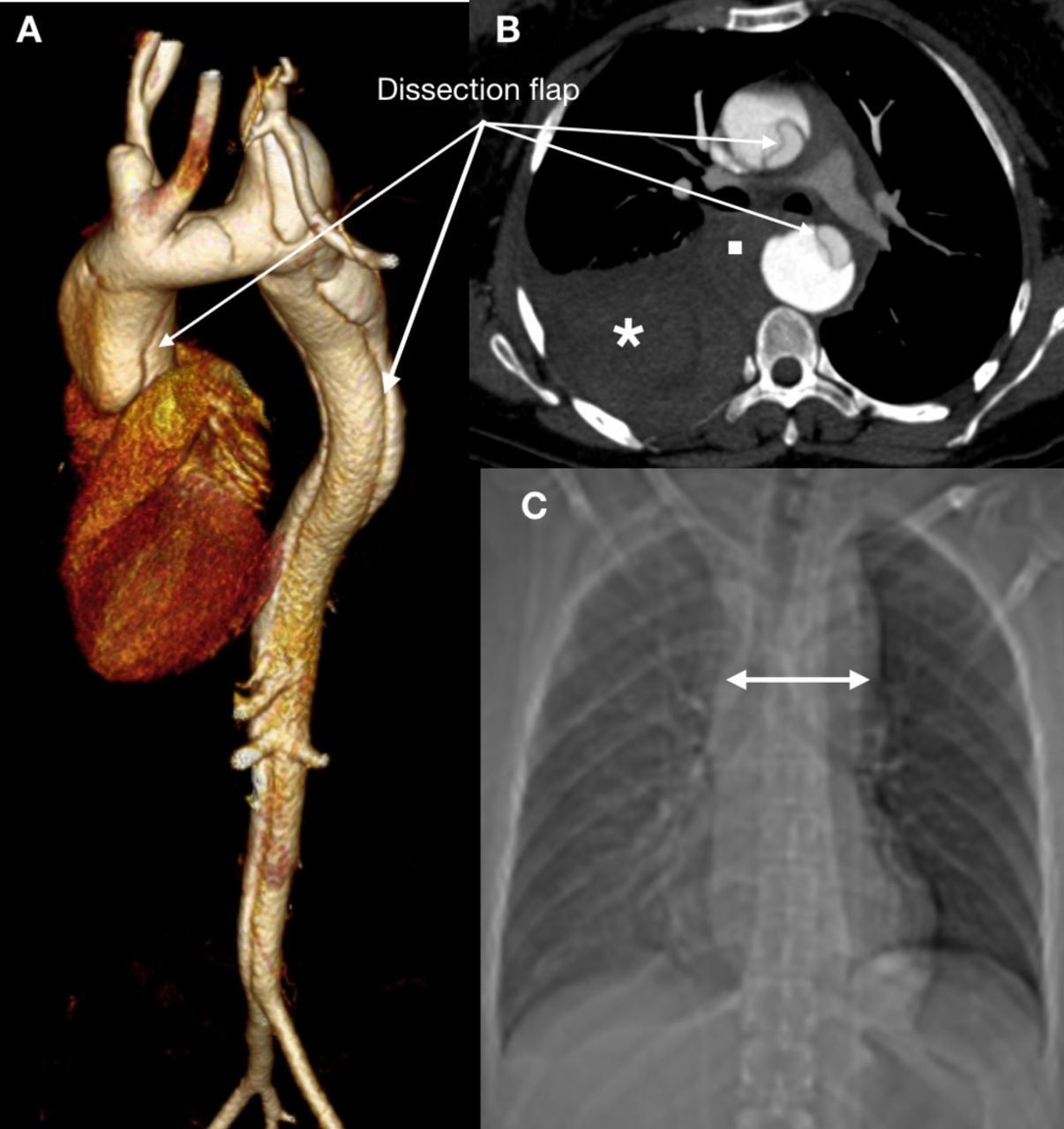
Acute aortic events are a major cause of morbidity and mortality in the young and middle-aged adult with TS (figure 2). The risk of aortic dissection is 20-fold to 100-fold increased compared with non-TS females, and dissection occurs in 1–2%.29 30 Reflecting an early-onset acquired and/or a hitherto undefined congenital intrinsic aortic wall defect, the median age at dissection is as young as 35 years.30 Dissections are mainly Stanford Type A (90%) that always affect the ascending aorta and/or aortic arch and may extend further distally. The remaining 10% are Stanford type B dissections that involve the descending aorta but never the more proximal aorta.18 29 30

Acute aortic syndrome occurs even in young and middle-aged adults with Turner syndrome (TS). This potentially fatal event is a concern in any adult woman with TS who presents with acute onset chest pain, and the threshold for performance of definitive imaging should be low. Definitive imaging will, depending on the clinical state of the patient as well as local practices and experience, involve cardiovascular CT (CCT), cardiovascular magnetic resonance imaging and/or transoesophageal echocardiography. CCT is often the preferred imaging modality owing to the minimally invasive nature, rapid image acquisition, diagnostic accuracy and wide availability. Here, CCT shows an acute aortic dissection in a 35-year-old woman with TS. The dissection flap of the Stanford Type A dissection extends from the aortic sinuses to the iliac bifurcation as is shown on a 3D volume-rendered reconstruction of the thoracoabdominal aorta (A). There is haemomediastinum (B, square) and haemothorax (B, asterisk) with mediastinal widening seen on the X-ray (C, double-headed arrow). There was an unobstructed but elongated transverse arch with ’bovine' branching, an aneurysm of the left subclavian artery and a bicuspid aortic valve. The dissection was treated urgently with an interposition graft to the ascending aorta.
Risk factors for aortic dissection in TS are aortic dilation, BAV, CoA, hypertension, karyotype 45,X and pregnancy.17 18 29 31 Imaging takes centre stage in risk stratification by diagnosing and monitoring the cardiovascular risk factors. Aortic dilation may occur anywhere in the thoracic aorta, but is most frequently encountered in the ascending aorta and when other aortic risk factors are present.15 16 22 32 A small physical stature in TS makes translation of absolute diameter thresholds from the general population less valuable, and indexation of aortic diameter to body surface area (BSA) appears to improve the predictive value for aortic dissection in TS.29 The approach of BSA-indexation is less well suited in children (< 15 years) where TS-specific z-scores are preferred. 33 34 Z-scores show how many SDs an absolute measurement is below or above the population mean.
Aortic surgical thresholds in TS are based on a few retrospective and case studies.17 18 29 The proposed thresholds are expert consensus, and they apply only to the mid-ascending aorta (at the level of the right pulmonary artery) with no evidence available for other aortic segments6 7 The risk at aortic surgery is undefined. Prophylactic aortic surgery is suggested at a mid-ascending aortic diameter ≥2.5 cm/m2 (BSA indexed) in adults (≥15 years). This recommendation is stronger when other risk factors are present. A very small or large BSA for TS (average BSA for TS is around 1.6 m2) skews indexed diameters, and here a raw threshold of 4.0 cm applies.6 ,7 33 . In children (<15 years), the surgical threshold is at a TS-specific z-score ≥4. Aortic growth >0.5 cm/year (or TS-specific z-score >0.5/year) is considered aggressive disease, which compares with average growth rates of 0.1–0.4 mm/year in adults with TS6 7 34 Aortic size also guides medical antihypertensive treatment and lifestyle advice, as detailed in the recent guidelines6 7 Aortic wall function seems perturbed in TS, but functional markers await validation for risk stratification.35 36
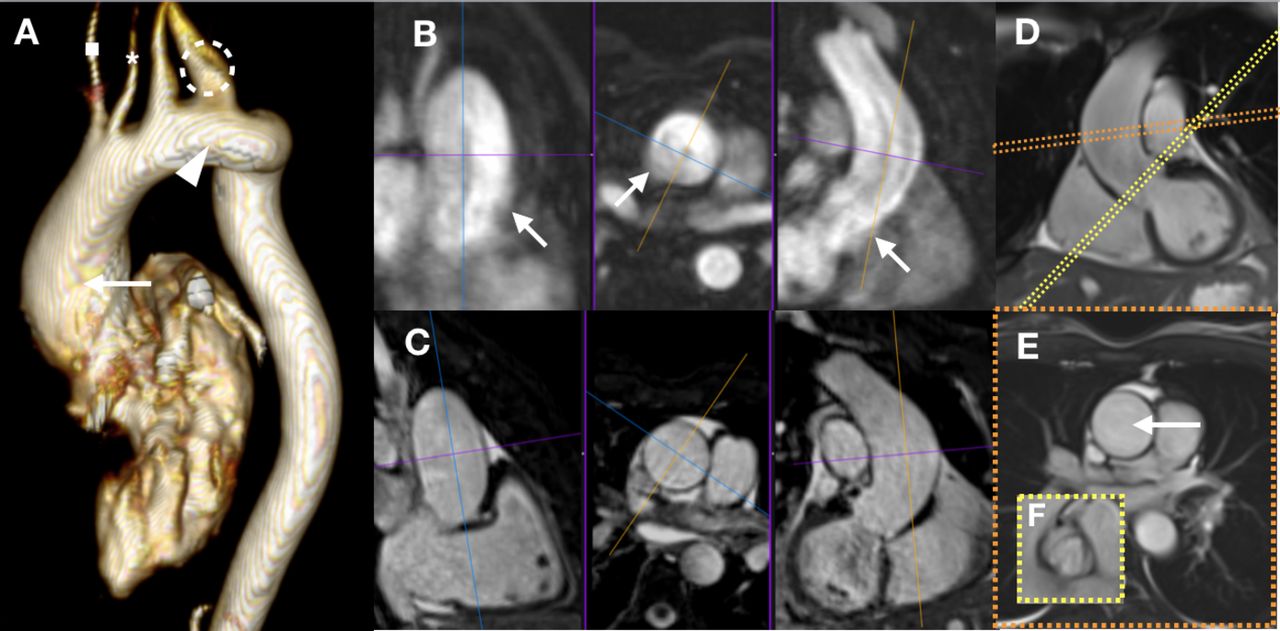
TTE generally suffices for aortic risk stratification in children. However, cross-sectional imaging becomes necessary in older teenagers and adults when acoustic windows become limited and aortic disease more likely (table 2).32 37 Aortic disease may involve any part of the thoracic aorta, and TTE is less sensitive beyond the aortic sinuses.32 38 CMR visualises all thoracic aortic segments, and 3D images can be reformatted for measurement of truly orthogonal diameters (’double-oblique') for optimal reproducibility (figure 3).32 39 CCT has high spatial resolution but radiation exposure, especially with serial surveillance, is a concern despite lower radiation doses on newer scanners. There is no consensus on the optimal cardiac phase for aortic diameter measurements. Systolic diameters may better reflect dissection risk, but diastolic diameters may be more reproducible.40 41 The principal outcome study for aortic diameters in TS used non-ECG-gated data.29 Irrespective of the evidence available, it is essential to acquire data in the same cardiac phase particularly for the ascending aorta and in serial imaging studies (figure 3). The aortic arch and descending aorta are less affected by motion so correlation with the cardiac phase is less imperative. Endoluminal measurements (inner edge-to-inner edge—excluding the aortic wall) are obtainable with all modalities and are preferred because they have formed the basis for comparative studies in TS.20 The same measurement technique should always be used in serial studies, and measurement variability will be at least 1–3 mm even with 3D ’double-oblique' measurements.20

Evaluation of aortic dissection risk by imaging in Turner syndrome (TS) includes non-invasive evaluation of the aortic valve, aortic arch phenotype and thoracic aortic diameter. A combination of transthoracic echocardiography and cardiovascular magnetic resonance (CMR) is often needed with cardiovascular CT performed in select girls and women. The addition of CMR is increasingly needed with age, as acoustic windows often become limited and the entire thoracic aorta must be assessed. Accurate aortic size measurements require ECG-gated imaging, especially for the ascending aorta, and 3D data with multiplanar reconstructions or dedicated 2D ‘double-oblique’ imaging in order to optimise measurement variability. Here, different CMR sequences are shown for a 35-year-old woman with TS under consideration for prophylactic aortic surgery. A 3D volume-rendered reconstruction of a contrast-enhanced, non-ECG-gated aortogram (A) shows: (1) a left-sided aortic arch with incidental separate arch origins of the right common carotid (A, asterisk) and right subclavian (A, square) arteries, (2) a dilated origin of the left subclavian artery (A, circle) with less dilated origin of the preceeding left common carotid artery, (3) an elongated and mildly tortuous but unobstructed aortic arch (A, arrowhead), and (4) the point of maximum dilation in the mid-ascending aorta (A, arrow). The same 3D data set allows multiplanar reconstructions for orthogonal diameter measurements at any point (B). However, the non-ECG-gated nature of the aortogram causes blurring of the aortic wall from cardiac motion leading to inaccurate measurement (B, arrows); this is less pronounced for the descending aorta. Data acquisition for a particular cardiac phase minimises any such blurring as seen in a multiplanar reformat of a non-contrast-enhanced ECG-triggered 3D volumetric data set (C). A specific cardiac phase can also be assessed with 2D imaging as is shown for an ECG-gated cine that is carefully positioned for the correct image plane (D, orange slice position) giving an orthogonal plane through the point of maximum aortic diameter (E with arrow indicating the mid-ascending aorta). The same principle is used for en face visualisation of the bicuspid aortic valve (D, yellow slice position giving the orthogonal plane (F)). The peak-systolic mid-ascending aortic diameter was 2.6 cm/m2, and the recommendation was for prophylactic aortic surgery from the multidisciplinary team at a specialist centre for adults with congenital heart disease.
Aortic diameters should be assessed for all females with TS. Imaging should include the proximal head and neck arteries. Surveillance frequencies in established aortic disease should be adapted pragmatically according to age, blood pressure, aortic diameter, aortic growth and the presence of congenital lesions such as CoA and BAV (figure 4)6 7 Aortic dissection may, however, also occur without conventional risk factors present, and the thoracic aorta should be reassessed every 5–10 years even when the initial aortic diameters are normal (table 3, figure 4)6 7 Strategies for imaging in suspected aortic dissection are the same as in the general population.40 42 However, due to the high risk of aortic dissection, definitive imaging such as CCT should be considered even in young women with TS when there are suggestive symptoms (figure 2).
- Download figure
- Open in new tab
- Download powerpoint
Imaging follow-up for aortic dilation in girls and women with Turner syndrome (TS) should be adapted to age, aortic size and aortic risk factors as per expert consensus in the international guidelines6 7 Risk factors include bicuspid aortic valve (BAV), aortic coarctation (CoA) and hypertension as diagnosed at the initial cardiovascular screening. This initial screening includes transthoracic echocardiography (TTE), cardiovascular magnetic resonance (CMR) and ambulatory blood pressure measurement. The aortic size criteria apply to the mid-ascending aorta at the level of the right pulmonary artery, and this size is given as the aortic diameter indexed to body surface area (ASI) in adult women and TS-specific z-scores (TSZ) in girls. No evidence exists to guide similar thresholds for other aortic segments in TS. The colours signify perceived risk: green is low, yellow is moderate and red is high. The addition of CMR to TTE should be determined by the quality of the acoustic windows, and remembering that TTE is less accurate for aortic disease that extends beyond the aortic sinuses when compared with CMR. CMR is more accurate than TTE owing to this modality being independent of standard imaging planes. If CMR is contraindicated or not feasible, then cardiovascular CT may be used as an alternative, but the associated radiation exposure should be considered.
Pregnancy and the postpartum state are risk factors for aortic dissection.31 TTE and CMR should be performed <2 years before a planned pregnancy, and CCT may be used when CMR is not possible (table 3)6 7 Pregnancy is discouraged when the mid-ascending aorta measures either ≥2.5 cm/m2 in isolation or ≥2.0 cm/m2 with other dissection risk factors present. Again, thresholds apply to the mid-ascending aorta only. During pregnancy and the postpartum period, TTE is advisable every 4–6 weeks when the mid-ascending aortic diameter is ≥2.0 cm/m2. CMR should be considered when aortic disease extends beyond the aortic sinuses. CMR without gadolinium is considered safe but still avoided by some, especially during the first trimester.43 The use of gadolinium during pregnancy is controversial and should be based on an individual risk–benefit analysis. Pregnant women with smaller aortic size and no other risk factor should have at least one TTE at 20 weeks of gestation, and renewed aortic imaging is needed in the postpartum period for all women with TS (table 3). Aortic diameters and growth inform obstetricians regarding optimal time and mode of delivery6 7 Pregnancy in the context of other CHDs is dealt with according to general population guidelines, but TTE every 6–12 weeks seems reasonable.
Cardiac shunts
Partial anomalous pulmonary venous connections (PAPVCs) to the systemic venous return are common (13–25%) (figure 5).4 44 45 This anomaly often associates with sinus venosus defects in the general population, but this has not been confirmed in TS. Total anomalous pulmonary venous connections are uncommon in TS. The volume loading incurred by the left-to-right shunt in PAPVCs may adversely impact the right ventricle as indicated by ventricular dilatation, leftward bowing of the interventricular septum in diastole and functional impairment. Long-standing overcirculation can lead to pulmonary arterial hypertension. Surgical correction follows general population guidelines and is considered when there is important right ventricular volume loading.24 25 In pulmonary arterial hypertension, surgical corrections should be determined on a case-by-case basis. Ventricular (1–5%) and atrial (1–2%) septal defects and patent arterial ducts may also cause shunting, but it is unclear if these occur more frequently as isolated findings in TS.5 CMR or CCT is often preferred in PAPVCs due to the comprehensive 3D anatomical assessment, which for CMR is combined with key prognostic markers including right ventricular volumes and function along with the shunt fraction that is calculated as the ratio of pulmonary blood flow relative to the systemic blood flow (Qp:Qs ratio). TTE is less sensitive than CMR for location of suspected PAPVCs and right ventricular assessment, and TTE cannot reliably quantify the Qp:Qs ratio. CCT provides high spatial resolution 3D angiography but cannot directly measure the Qp:Qs ratio, and ventricular volumetric analysis by retrospective ECG-gated CCT incurs a relatively high radiation dosage despite radiation dose improvements on newer scanners. If surgical correction is not warranted, follow-up imaging should be individualised according to symptoms, shunt fraction, and right ventricular volumes and function.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Partial anomalous pulmonary venous connections in Turner syndrome (TS) may involve the pulmonary venous return of either lung. Frequently, the right upper and middle lobe pulmonary veins connect to the superior vena cava (SVC) and/or the left upper lobe connects to the innominate vein. Here, a cardiovascular magnetic resonance study in a 48-year-old woman with TS shows the pulmonary veins of the right upper and middle lung lobes connecting anomalously (A, arrows) to the SVC. The anomalous entry point is indicated (A, circle). The SVC is single, right-sided and drains to the right atrium (RA). The right ventricle (RV), aorta (AO) and main pulmonary artery (PA) are also seen on this 3D volume-rendered reformat of the contrast-enhanced non-ECG-gated angiography. Dedicated ECG-gated cine views of the left (B) and right (C) ventricular outflow tracts can be used to plan image positions for direct measurement of aortic and pulmonary flows. The orange imaging plane (B) indicates the plane for the aortic flow with the phase contrast flow data shown to the right and the AO indicated (arrowhead). Similarly, the yellow imaging plane (C) corresponds to the plane of the pulmonary phase contrast flow data as shown to the right with the PA indicated (arrowhead). These image positions can also be derived from the 3D angiographic data set. The phase contrast flow data sets are used to calculate the ratio between systemic and pulmonary flow (Qp:Qs ratio). Further cine imaging is acquired, including the four-chamber view (D) that is used to plan a short-axis stack of ECG-gated cine images. This stack is then used for quantitative analysis of ventricular volumes and function. In this woman, there were signs of significant right heart volume loading: the RV was dilated (RV to left ventricular ratio of end-diastolic volumes was 1.7:1), the interventricular septum was flattened in diastole and there was a left-to-right shunt with high pulmonary blood flow as evident by a Qp:Qs of 1.8:1. Surgical correction was performed after multidisciplinary team discussion at a specialist centre for adults with congenital heart disease.
Coronary arterial disease
Anomalies of coronary arterial origin and course are frequently encountered in TS (20%).46 However, the vast majority are incidental and benign and they may only become significant when planning cardiothoracic surgery.12 21 46 ECG-gated coronary CCT has excellent negative predictive value for both congenital and acquired coronary artery diseases (table 2). TTE and CMR will, however, often suffice in children, adolescents and young adults when coronary atherosclerosis is not a concern. Conversely, the threshold should be low for targeted imaging for obstructive coronary artery disease in adult women with TS who are at increased risk of early-onset ischaemic heart disease.1 2 47 No age-specific cut-offs exist for imaging of coronary atherosclerosis in asymptomatic females with TS prior to aortic surgery or valve replacement. However, CCT is often performed in asymptomatic adults with TS before surgery due to the non-invasive nature and a relatively low radiation dose (table 2).
Myocardial disease
TS is not known to be an independent risk factor for cardiomyopathy. However, left ventricular diastolic dysfunction is often seen and may be accompanied by an increase in myocardial mass.48 49 These abnormalities may occur in the presence of frequent systemic arterial hypertension, aortic valve dysfunction and/or aortic arch anomalies. However, asymptomatic young females without risk factors may also have left ventricular disease with abnormal diastolic function and, possibly, abnormal myocardial metabolism.48–50 Isolated left ventricular systolic dysfunction is uncommon. Endocardial fibroelastosis can occur in young girls with TS, especially in the context of severe left ventricular outflow tract obstruction. Other congenital myocardial anomalies such as non-compaction are rare. TTE is the principal clinical imaging modality, but CMR is the reference standard for left ventricular mass, volumes and systolic function, and for myocardial tissue characterisation.
Conclusion
Cardiovascular imaging is essential to providing excellent clinical care to girls and women with TS in order to alleviate the negative impact of congenital and acquired CVDs and to reduce morbidity and mortality. Cardiovascular imaging should be regularly used in all patients, irrespective of karyotype and phenotype, owing to a high incidence of cardiovascular lesions that are often subclinical, may determine outcomes and occur in patterns specific to TS. A pragmatic approach is recommended to the application of cardiovascular imaging due to limited knowledge of both the natural history of CVD and outcomes with different cardiovascular interventions in girls and women with TS.
References
Footnotes
Contributors All authors have contributed to the drafting of the manuscript, and all authors have approved the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Correction notice Since this article was first published online figure 4 has been updated.