Article Text

Abstract
Objectives Patients with rheumatoid arthritis (RA) display an increased risk of heart failure independent of traditional cardiovascular risk factors. To elucidate myocardial disease in RA, we have investigated molecular and cellular remodelling of the heart in an established mouse model of RA.
Methods The collagen antibody-induced arthritis (CAIA) RA mouse model is characterised by joint inflammation and increased inflammatory markers in the serum. We used CAIA mice in the postinflammatory phase that resembles medically controlled RA or RA in remission. Hearts were collected for cardiomyocyte isolation, biochemistry and histology analysis.
Results Hearts from mice subjected to CAIA displayed hypertrophy (heart/body weight, mean±SD: 5.9±0.8vs 5.1±0.7 mg/g, p<0.05), fibrosis and reduced left ventricular fractional shortening compared with control. Cardiomyocytes from CAIA mice showed reduced cytosolic [Ca2+]i transient amplitudes (F/F0, mean±SD: 3.0±1.2vs 3.6±1.5, p<0.05) that was linked to reductions in sarcoplasmic reticulum (SR) Ca2+ store (F/F0, mean±SD: 3.5±1.3vs 4.4±1.3, p<0.01) measured with Ca2+ imaging. This was associated to lower fractional shortening in the cardiomyocytes from the CAIA mice (%FS, mean±SD: 3.4±2.2 vs 4.6%±2.3%, p<0.05). Ca2+ handling proteins displayed oxidation-dependent posttranslational modifications that together with an increase in superoxide dismutase expression indicate a cell environment with oxidative stress.
Conclusions This study shows that inflammation during active RA has long-term consequences on molecular remodelling and contractile function of the heart, which further supports that rheumatology patients should be followed for development of heart failure.
- heart disease
- heart failure
- translational cardiovascular science
- myocardial disease basic science
- cardiac risk factors and prevention
Statistics from Altmetric.com
- heart disease
- heart failure
- translational cardiovascular science
- myocardial disease basic science
- cardiac risk factors and prevention
Introduction
Rheumatoid arthritis (RA) is a systemic inflammatory syndrome that predominantly affects the joints and can also manifest in multiple extra-articular organs. Patients with RA have increased risk of developing heart disease from both ischaemic and non-ischaemic causes, and these are major contributors to mortality and disease burden in RA.1 Myocardial contractile activity seems particularly vulnerable to systemic inflammation, as patients with RA have higher risk of developing heart failure that is not explained by traditional cardiovascular risk factors.2 We recently showed that patients with RA have increased incidence of non-ischaemic heart failure generally, in the early stages of RA and in correlation to serum inflammatory markers.2 This indicates that the systemic inflammation in RA has the capacity to impair cardiac function and sensitise the heart to develop heart failure independent of ischaemic heart disease.
Cardiomyocyte contractile function is graded by free cytosolic [Ca2+] ([Ca2+]i), which is controlled by diverse regulatory factors including redox-dependent modifications of Ca2+ handling proteins.3 On activation, the [Ca2+]i increases transiently in cardiomyocytes via opening of the L-type Ca2+ channel (dihydropyridine receptor (DHPR)) and sarcoplasmic reticulum Ca2+ release channels, the ryanodine receptor 2 (RyR2). Opening of RyR2 dissipates the [Ca2+]i gradient from the SR storage to the cytoplasm. The capacity to release Ca2+ is highly dependent on the Ca2+ stored within the SR,4 which is regulated by the Ca2+ buffer protein calsequestrin 2 (CSQ2). The RyR2 is a target for reactive oxygen and nitrogen stress that can cause redox-dependent modifications of amino acid residues (eg, tyrosine nitration, cysteine nitrosylation, malondialdehyde adducts and carbonylation/oxidation) that adversely influence the SR Ca2+ release.5
Oxidative stress plays an important role in chronic inflammatory conditions such as RA. Increased production of reactive oxygen species (ROS) during inflammation causes DNA mutations, lipid peroxidation and protein oxidation, leading to impaired cell function.6 Markers of oxidative damage have been found in the synovia and in serum that correlate to the disease activity in RA.7 In heart failure, ischaemic heart disease and systemic inflammation, ROS-mediated signals have been implicated in maladaptation of excitation–contraction coupling (eg, Ca2+ handling and myofilament function).3 8
Molecular studies of myocardial remodelling and cardiomyocyte contractile signalling have, to our knowledge, never been studied in the context of RA. Thus, the aim of this study was to investigate if inflammation during active RA has long-term consequences on remodelling and contractile function of the heart. For this purpose, we used the collagen antibody-induced arthritis (CAIA) mouse model that, similar to patients with RA, present with joint inflammation, elevated inflammatory markers in serum during the acute phase9 (online supplementary figure S1) and changes in, for example, sensory thresholds, bone structure and neurochemical profile that outlast the inflammation by many weeks. We examined mice subjected to CAIA in the ‘late phase’ approximately 1–2 months after the signs of inflammatory arthritis had resolved, and animals had entered a state that resembles medically controlled RA or RA in remission. Hearts from CAIA and control (CTR) mice were used to characterise morphological, molecular and functional differences at the level of the whole heart and in preparations of isolated cardiomyocytes (figure 1). Cardiovascular comorbid conditions in RA are common and are increasingly acknowledged to present therapeutic challenges in the intersection of cardiology and rheumatology.10 Our results show changes in cardiac remodelling and function that provides important insight to the link between RA and sensitisation to develop heart failure.
Supplementary file 1
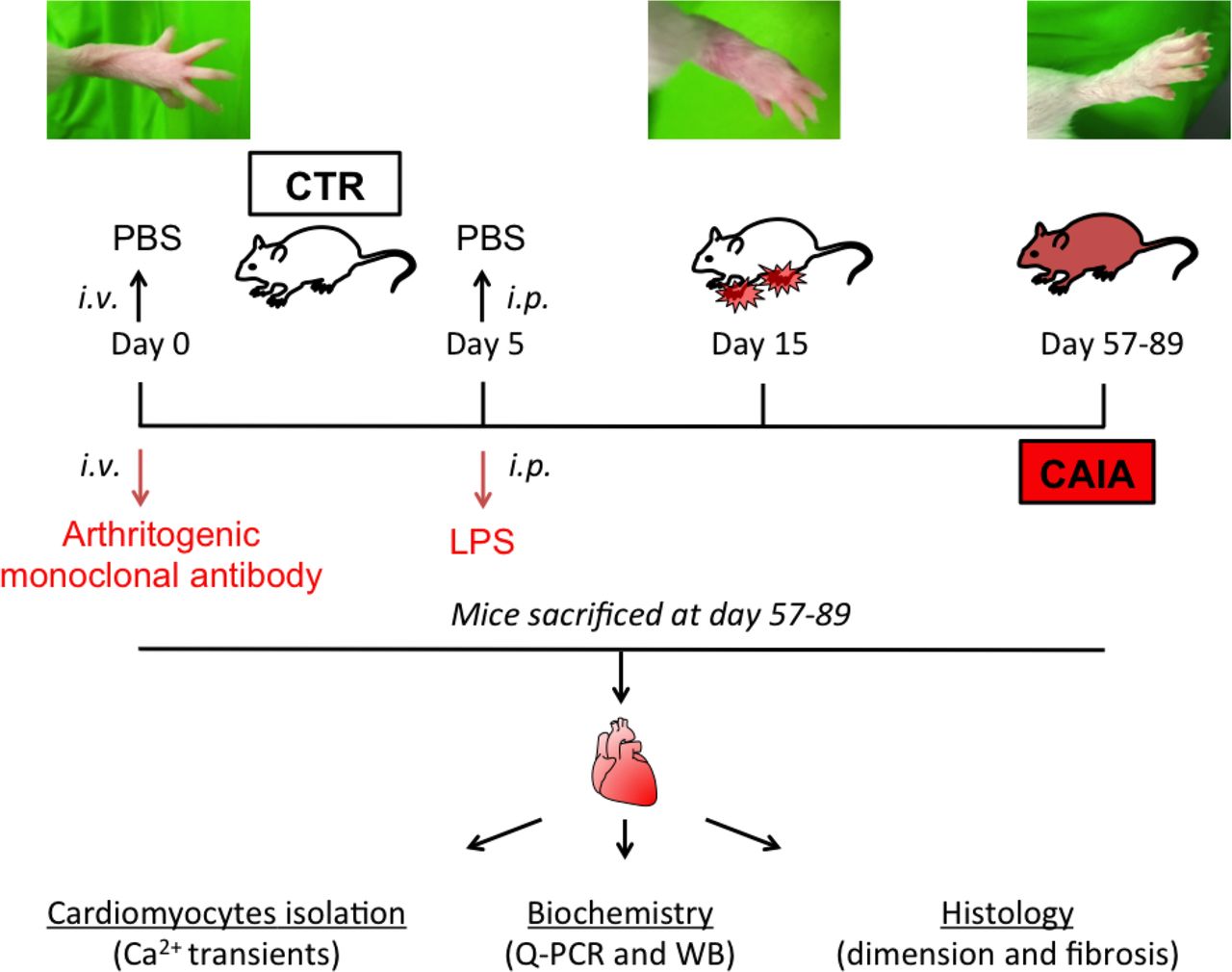
Collagen antibody-induced arthritis (CAIA) model. On day 0, mice were injected intravenously (iv) with an anti-collagen type II arthritogenic cocktail containing five monoclonal antibodies. After 5 days, 25 µg of lipopolysaccharide (LPS) was injected intraperitoneally (ip) to synchronise and enhance the inflammatory response. The control group (CTR) received iv phosphate-buffered saline (PBS) on day 0 and saline ip on day 5. The joint inflammation was monitored by visual inspection and peaked around day 15 after antibody injection, and then gradually decreased. We used animals in the postinflammatory phase (57–89 days after induction), which resembles medically controlled RA or RA in remission. Animals underwent echocardiography and were then sacrificed. Hearts were collected for cardiomyocyte isolation, biochemistry (eg, western immunoblotting; WB) and histology. RA, rheumatoid arthritis.
Methods
Measurement of [Ca2+]i with Fluo-3 AM
Cardiomyocytes were loaded with the fluorescent indicator Fluo-3 AM (~5 µM; Invitrogen) for ~20 min. [Ca2+]i transients were measured using a confocal microscope (Biorad) in line scan mode.11 A wavelength of 491 nm was used for excitation, and emitted light was collected at wavelength >515 nm. The line scan was oriented along the long axis of the cardiomyocyte. The fluorescent signal (F/F0) was calculated as the ratio between resting fluorescence before the start of the [Ca2+]i transient (F0) and the peak fluorescence (F) of the [Ca2+]i transient. In some experiments, cells were exposed to the β-receptor agonist isoproterenol (ISO; 100 nM in Tyrode solution) or caffeine (10 mM in Tyrode solution) for Ca2+ imaging at maximum inotropy or to measure the SR Ca2+ stores, respectively.
Detailed methods are described in the online supplementary material.
Supplementary file 2
Results
Heart remodelling in RA mice
We first investigated if the CAIA mice displayed heart remodelling. Formalin-fixated hearts were cross-sectioned, and cardiac ventricular dimensions were measured. Moreover, heart dimensions were measured in vivo using echocardiography. The CAIA group displayed larger ventricular dimensions, septum width and left ventricular diameter (figure 2A, Supplementary table S1). The mRNA expression of atrial natriuretic factor, brain natriuretic peptide and beta-myosin heavy chain (β-MHC) was increased in CAIA group compared with control indicating hypertrophic signalling (figure 2B). Moreover, the heart weight–body weight ratio was higher in CAIA compared with control mice (figure 2C). To further study myocardial remodelling, we measured expression of genes involved in hypertrophy signalling. To test for maladaptive remodelling, we measured expression of genes involved in fibrosis (collagen 1 and transforming growth factor beta 1, Tgf-β1) and investigated the level of tissue fibrosis by staining histological sections of cardiac ventricle with Masson’s trichrome staining. Compared with control, the hearts from CAIA mice displayed higher degree of fibrosis positive staining (figure 2D,E), which together with increased mRNA expression of collagen 1 and Tgf-β1 (figure 2F), show that the CAIA mice display maladaptive heart remodelling. The proinflammatory cytokine tumour necrosis factor (TNF) is a key mediator in the pathogenesis of RA that is known to be expressed both in serum and in the heart.12 The concentration of TNF was increased in the CAIA mouse serum during the peak arthritis phase (online supplementary figure S1B). Interestingly, the cardiac mRNA expression of TNF was increased also in the late phase CAIA mice when the overt signs of arthritis were gone (figure 2F). This mirrors the local cytokine activity that is found in the myocardium of patients in chronic RA.12 Furthermore, we measured the inflammatory mediator IL1β in serum and heart tissue without seeing any difference between CAIA and control mice (online supplementary figure S1C,D). In summary, these data indicate that hearts from the RA mouse model CAIA display molecular and morphological remodelling.
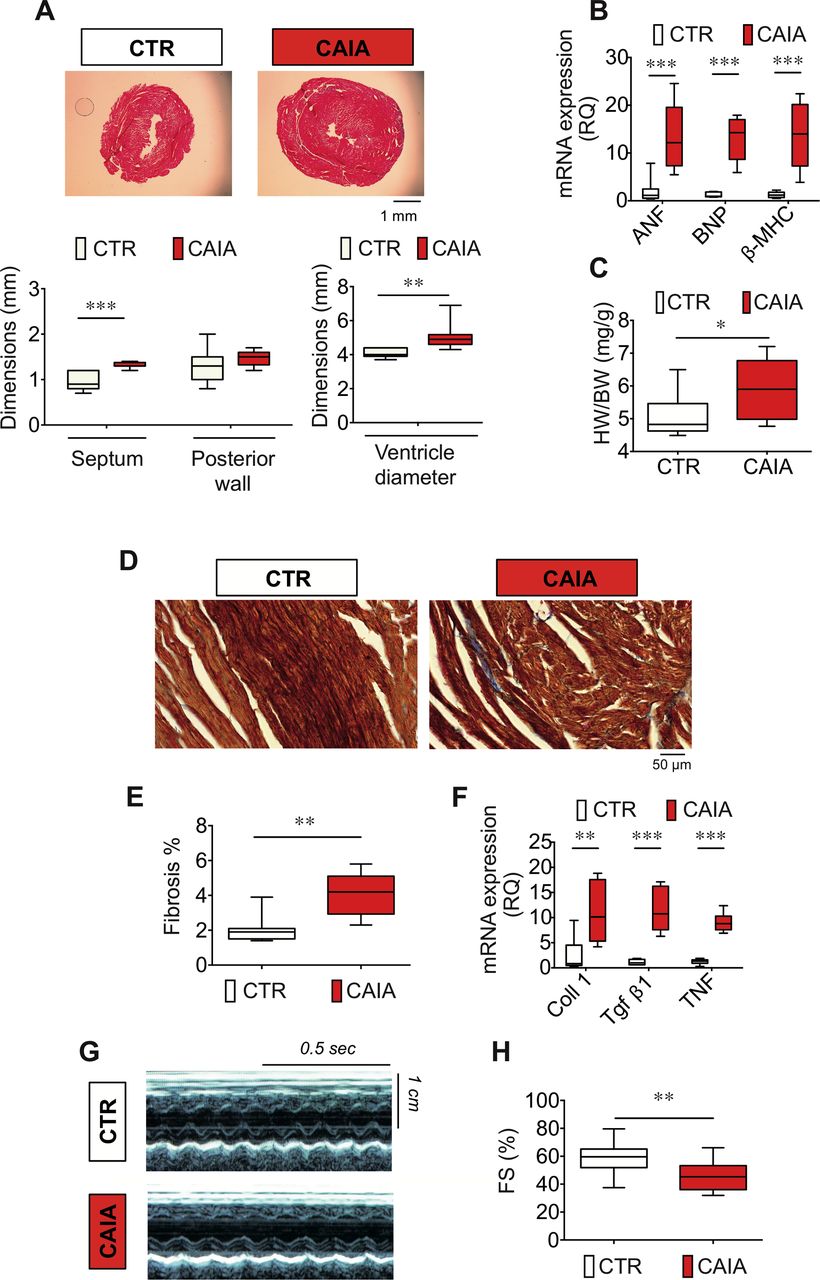
Structural remodelling and contractile dysfunction in hearts of CAIA mice. Representative H&E stained cardiac ventricular cross-sections (A, top) and heart dimensions of CAIA mice displaying cardiac hypertrophy with increased septum width and left ventricular diameter (n=7–8; A, bottom). The expression of genes involved in ventricular remodelling (ANF, BNP and β-MHC) was increased in hearts of CAIA mice (values expressed as mRNA expression relative quantification (RQ), (n=7; B), and increased heart weight–body weight ratio (HW/BW), n=10–12; C). Representative cardiac ventricular cross-sections with Masson’s trichrome staining (indicated in blue) showing fibrotic depositions (40× magnification objective, scale bar indicates 50 µm; D). Quantification of interstitial cardiac fibrosis (expressed as % fibrosis, n=8–9; E). mRNA expression of genes involved in fibrogenesis remodelling and myocardial mRNA expression of the inflammatory cytokine TNF in CAIA and control mice (collagen 1 (Coll 1), Tgf-β1 and TNF; values expressed as mRNA expression RQ, n=7; F). Transthoracic echocardiography measurement of fractional shortening (FS%) calculated from left ventricular M-mode parameters (FS%=LVd−LVs/LVd*100) in CAIA and control mice (G and H). Representative images and average data (n=15) and are displayed in figure parts G and H, respectively. Data are displayed as box plots. *p<0.05, **p<0.01, ***p<0.001. ANF, atrial nartiuretic factor BNP, brain natriuretic peptide; CAIA, collagen antibody-induced arthritis; CTR, control; TNF, tumour necrosis factor; β-MHC, beta-myosin heavy chain; Tgf-β1, transforming growth factor beta 1.
Lower fractional shortening in CAIA measured by echocardiography
To evaluate the cardiac function and morphology in vivo, we used transthoracic echocardiography. Left ventricular fractional shortening (FS%) was slightly reduced in the CAIA compared with control mice (figure 2G,H). Heart rate, measured as the contraction frequency on the echocardiogram showed no significant difference between the groups (beat per minute, mean±SD CTR: 490±55 vs CAIA 460±68, p=0.2).
Reduced [Ca2+]i transients and contractility in cardiomyocytes isolated from CAIA mice
To study if the arthritis mouse model displayed changes in cellular Ca2+ handling, we used isolated cardiomyocytes from CAIA and control (CTR) mice that were loaded with the fluorescent Ca2+ indicator Fluo-3. Compared with control, cardiomyocytes from CAIA mice displayed reduced [Ca2+]i transient amplitudes (figure 3A,B). Consistent with the lower [Ca2+]i transient amplitudes, fractional shortening was lower in cardiomyocytes isolated from CAIA compared with control animals (FS%, figure 3C). Moreover, the [Ca2+]i transient rate of increase was lower in the CAIA group (figure 3D). There was no significant difference between CTR and CAIA groups in the decay of [Ca2+]i transient as measured by the decay time constant (τ, figure 3E).

Impaired Ca2+ handling and contractility in cardiomyocyte isolated from CAIA mice. Measurement of cardiomyocyte [Ca2+]i transients with confocal microscopy and the fluorescent Ca2+ indicator Fluo-3 AM in line scan mode. Representative line scan images (A, top) and [Ca2+]i transient (A, bottom). Average [Ca2+]i transient amplitudes (F/F0, n=45–55 cells) show lower amplitudes in the CAIA (B). Fractional shortening, measured as the % change in cell length, was lower in CAIA compared with control group (FS%, n=43–55, (C). In figure parts D and E, [Ca2+]i transients kinetics are displayed. At basal condition, there was a significant difference in the average rate of increase (ROI) with CAIA displaying lower ROI (D). There was no difference in the rate of decay measured with the decay time constant (τ) (E). Data are displayed as box plots. *p<0.05. CAIA, collagen antibody-induced arthritis; CTR, control.
The β-adrenergic agonist isoproterenol is a potent inotropic stimulator that increases [Ca2+]i cycling in cardiomyocytes. In the presence of ISO (100 nM), no difference was found in CAIA and control for the amplitudes, ROI and decay rate of the [Ca2+]i transients nor for the cardiomyocyte fractional shortening (online supplementary figure 3A–D).
Supplementary file 3
Reduced L-type Ca2+ current in CAIA cardiomyocytes
The reduced [Ca2+]i amplitudes in cardiomyocytes isolated from CAIA mice could be due to decreased SR Ca2+ release and/or reduced Ca2+ entry on depolarisation. To test the latter, we measured the L-type Ca2+ current (ICa-L) using whole cell patch clamp technique on cardiomyocytes from control and CAIA mice. ICa-L was reduced over a range of voltage steps in the CAIA group compared with control (figure 4A,B). The peak ICa-L, measured at whatever voltage it occurred, was significantly smaller in the CAIA group (control 20.5±4.3 pA/pF, CAIA 13.1±2.9 pA/pF, n=7, p<0.01). The membrane potential (Em) for peak ICa-L was not significantly different (CTR, 0.1±2.0 mV, n=7; CAIA, 5.7±2.1 mV, n=7; p=0.07). Together, this shows that cardiomyocytes from CAIA mice display decreased Ca2+ entry during action potential activation but does not rule out a concomitantly altered SR Ca2+ release.

Reduced L-type Ca2+ current and SR Ca2+ storage in cardiomyocytes from CAIA mice. Whole cell patch clamp measurement of L-type Ca2+ current (ICa-L) was performed in cardiomyocytes from CAIA and control mice. Representative peak ICa-L at the potential indicated (A). Average I–V curve for ICa-L currents (mean±SEM, n=7) (B). CAIA and control cardiomyocytes loaded with Fluo-3 AM were superfused with caffeine (10 mM), which opens the RyR2 and empties Ca2+ from the SR. The resulting Fluo-3 fluorescent signal reflects the SR Ca2+ load. A significant difference in caffeine-induced SR unloading was seen between CAIA and control cardiomyocytes as displayed by representative traces (C) and average peak F/F0 (n=34–30) (D). Data are displayed as box plots. *p<0.05, **p<0.01. CAIA, collagen antibody-induced arthritis; CTR, control.
Measurement of SR Ca2+ stores
The reduced stimulation-induced [Ca2+]i could be explained by a reduced ICa-L. However, it is possible that RyR2-mediated SR Ca2+ release could be reduced as well. The SR Ca2+ release is highly dependent on the [Ca2+]i concentration gradient across the SR membrane. To test this, CAIA and control cardiomyocytes loaded with Fluo-3 AM were first paced in physiologic control buffer, then pacing was stopped and cells were superfused with caffeine (10 mM), which fully opens the RyR2 and unloads the Ca2+ from the SR. The peak of the caffeine-induced [Ca2+]i transient reflects the SR Ca2+ load. Compared with the control, cardiomyocytes from CAIA mice displayed reduced caffeine-induced [Ca2+]i transient amplitudes (figure 4C,D). In summary, our data suggest that the SR Ca2+ load is reduced in cardiomyocytes from CAIA animals, which could further explain the reduced cytosolic [Ca2+]i transients seen in heart of CAIA mice.
Post-translational modification of Ca2+ handling proteins in the CAIA heart
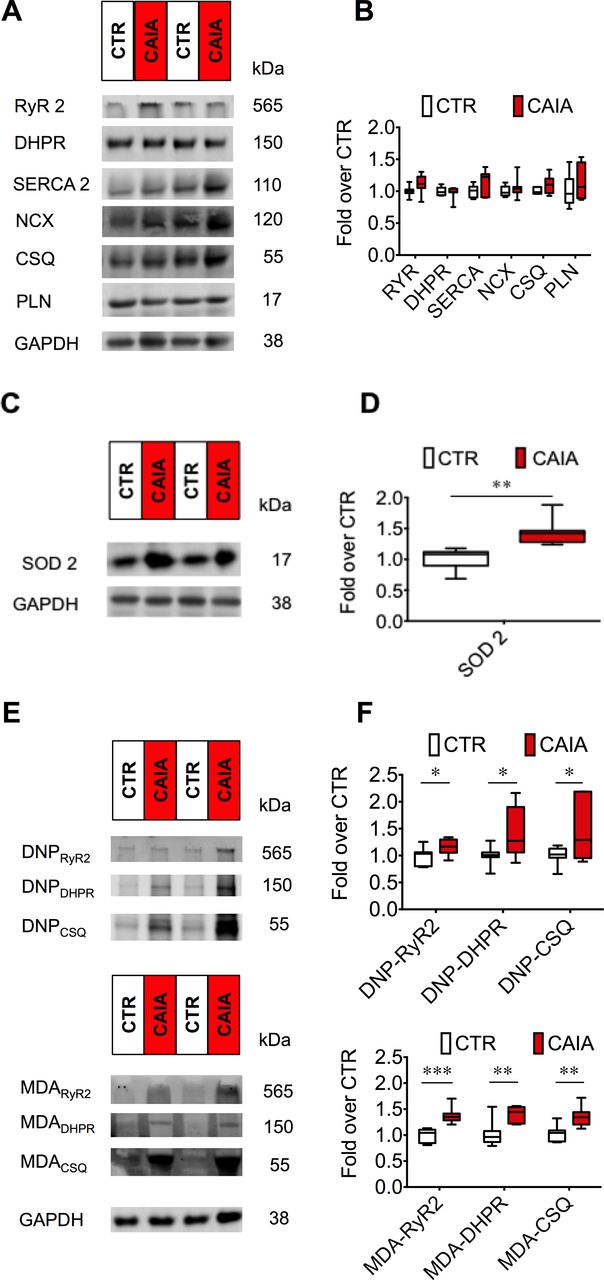
The protein levels of RyR2, DHPR, sarcoplasmic reticulum Ca2+ ATPase 2, Na+/Ca2+ exchanger, calsequestrin 2 (CSQ2) and phospholamban were not different in the hearts of CAIA and control mice (n=7, p>0.1; figure 5A,B). Thus, despite the reduction of contractility and Ca2+ transients seen in cardiomyocytes, the overall expression of Ca2+ handling proteins was not affected suggesting that other factors influence the Ca2+ signalling system.

Expression of Ca2+ handling proteins and ROS-dependent post-translational protein modifications. Cardiac protein expression measured with western immunoblotting for key Ca2+ handling proteins: ryanodine receptor 2 (RyR2), L-type Ca2+ channel/dihydropyridine receptor (DHPR), sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2), Na+/Ca2+ exchanger (NCX), calsequestrin 2 (CSQ2) and phospholamban (PLN). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as loading control. No difference was seen in the expression of above proteins in hearts of CAIA and control mice, representative western blots (A) and average data and (B) average data expressed as fold over control, n=7. Compared with control, the expression of the redox regulatory protein superoxide dismutase 2 (SOD2) was significantly higher in hearts of CAIA mice. Representative western blots (C) and average measurements (D). Post-translational modifications of proteins can form in conditions of oxidative stress. Oxidation-dependent modifications DNP (measures carbonylation) and the lipidperoxidation product malondialdehyde in hearts of CAIA and control mice. Representative western blots (E). Data quantification were displayed as box plots (F). *p<0.05, **p<0.01, ***p<0.001. CAIA, collagen antibody-induced arthritis; ROS, reactive oxygen species.
ROS can increase following inflammation and are known to cause protein oxidations that can impair cardiac function.3 In hearts of CAIA mice, the expression of the mitochondrial antioxidant protein SOD2 was increased (figure 5C,D). Increased expression of antioxidant enzymes is seen in response to increased ROS and oxidative stress. To investigate the presence of oxidative stress, we studied two oxidation-dependent protein modifications, carbonylation (DNP) and malondialdehyde (MDA) on the Ca2+ handling proteins, which have been shown to affect cardiac and skeletal muscle function in ageing and heart failure.3 8 Immunoblotting with antibodies against carbonylation or malondialdehyde modified proteins was used. In hearts of CAIA animals, RyR2, DHPR and CSQ2 displayed more MDA modifications as well as oxidation-dependent DNP modifications on the RyR2, DHPR and CSQ2 proteins compared with control group (figure 5E,F).
Discussion
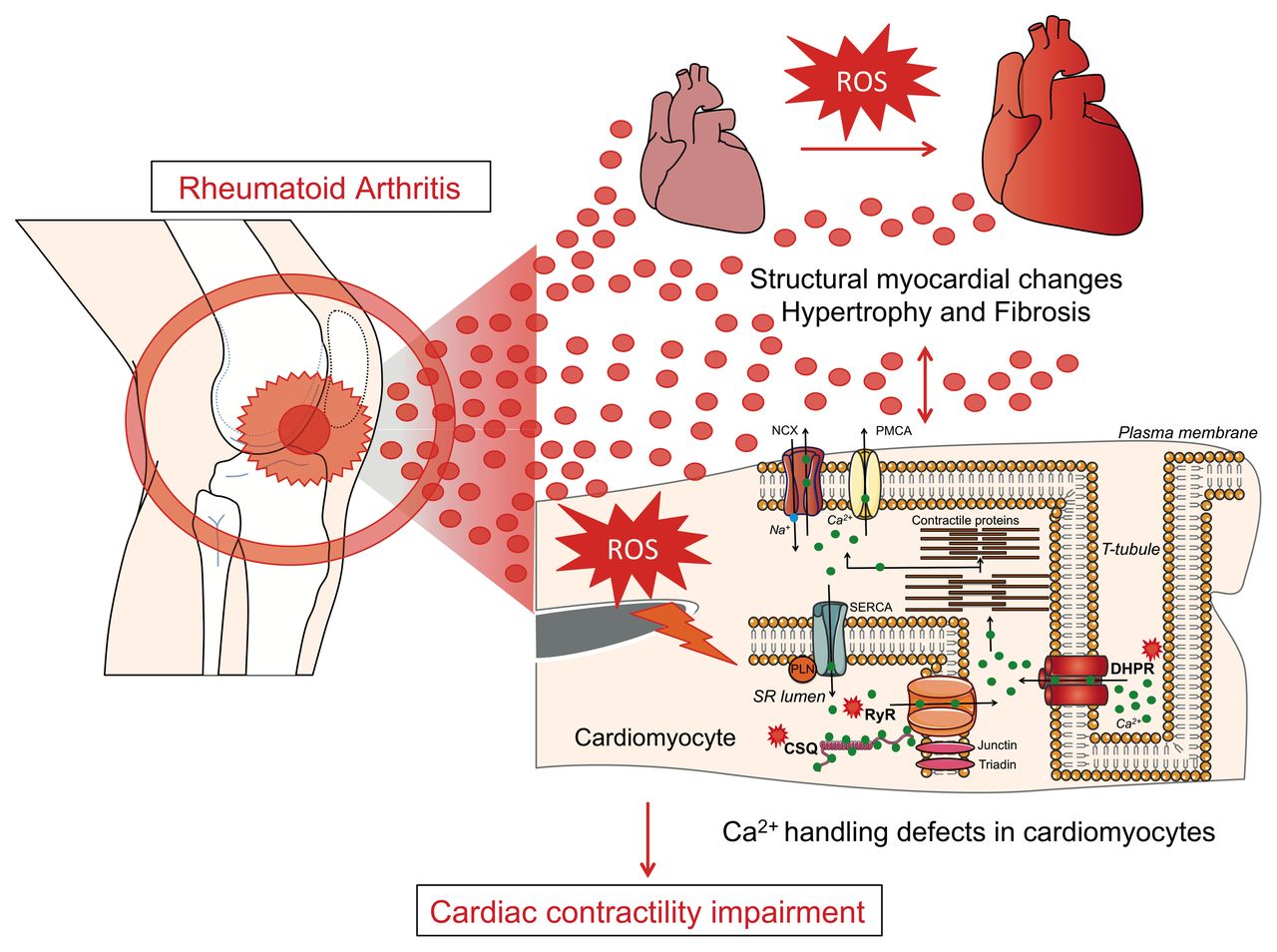
We show for the first time that the CAIA mouse model, which mimics human RA,13 displays myocardial remodelling consisting of ventricular hypertrophy, fibrosis, impaired cardiomyocyte Ca2+ handling and contractile dysfunction. This finding provides evidence for an acquired cardiomyopathy with reduced systolic and diastolic ventricular reserve function that would increase the susceptibility for development of overt heart failure (figure 6).

![[SP1.jpg]](https://heart.bmj.com/content/heartjnl/104/24/2026/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
![[SP3.jpg]](https://heart.bmj.com/content/heartjnl/104/24/2026/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
Cartoon depicting cardiac remodelling in experimental rheumatoid arthritis (RA). RA leads to structural and functional remodelling of the heart consisting of cardiac hypertrophy and fibrosis coupled to impaired contractile function. In cardiomyocytes, high levels of reactive oxygen species (ROS) promote post-translational modification of important Ca2+ handling proteins (RyR2, DHPR and CSQ). This was linked to impaired Ca2+ handing with reduced L-type Ca2+ current, SR Ca2+ stores and peak Ca2+ transient amplitudes and reduced cardiomyocyte contractility. CSQ, calsequestrin; DHPR, dihydropyridine receptor; RyR2, ryanodine receptor 2.
Systemic inflammation has been linked to oxidative stress in many cell types, including skeletal muscle.14 Protein carbonylation is a marker for oxidative damage that accumulates during ageing,8 in ischaemia/reperfusion and in diabetes. We found that hearts from mice subjected to CAIA display more oxidation-dependent modifications, including carbonylation, of cardiac Ca2+ handling proteins than hearts from control mice. Increase in oxidative protein modifications mirrors cellular oxidative stress that may be the consequence of increased ROS production or reduced oxidative defence. In our study, the latter is unlikely as there was an increased expression of the antioxidant defence enzyme SOD2, which instead indicates that the heart has responded to an oxidative stress environment.15
In heart failure and cardiomyopathy, ROS-mediated signals have been attributed a causative role in the disease progression. Redox-dependent post-translational modifications can impair protein function that leads to dysregulation of ion channels, altered Ca2+ handling and accelerated fibrosis.3 16 Moreover, oxidative stress signalling has been shown to activate pathways linked to hypertrophy remodelling in the heart. Clinical data suggest that cardiac structural remodelling is common in patients with RA even without overt heart failure.17 Multiple signalling pathways are involved in the development of cardiac hypertrophy, and ROS can stimulate some of these pathways.18 Our data show a cardiac hypertrophic phenotype in CAIA mice with increased expression of the β-myosin heavy chain gene, ventricular wall thickness and heart weight. Maladaptive cardiac hypertrophy commonly involves fibrotic deposition that disrupts the normal myocardial architecture and impairs the systolic and diastolic function of the heart. Tgf-β is a key signalling peptide that mediates fibrogenesis by activating cardiac fibroblasts, which promote deposition of extracellular matrix proteins such as collagen 1.19 We found that the hearts of mice subjected to CAIA display profibrogenic signalling as the gene expression of Tgf-β and collagen 1 was increased, which corroborates previous literature showing increased cardiac fibrosis and oxidative stress in a rat model of arthritis.20 Fibrogenesis signalling can be triggered by a range of stimuli with chronic inflammation being one typical stimulus. Indeed, inflammatory mediators, defective resolution of inflammation and ROS can result in sustained production of fibrogenic growth factors including Tgf-β.19 Intriguingly, CAIA mice displayed increased expression of the proinflammatory cytokine TNF locally in the heart that may act as a local driver for remodelling also in the absence of active systemic inflammation. In line with this, increased expression of cytokines in heart biopsies from patients with inflammatory rheumatic disease was not directly correlated with serum markers of inflammation.12 Furthermore, transgenic mice with heart-specific overexpression of TNF develop cardiomyopathy and heart failure, which supports a causal role of this cytokine in the pathogenesis of cardiac disease.21
Left ventricular fractional shortening was lower in CAIA mice than in control, although the impairment was slighter in magnitude than typically seen in mice with overt heart failure (eg, following myocardial infarction or chronic pressure overload).22 This finding of a mildly reduced ventricular function is consistent with RA patient data showing a slightly impaired myocardial systolic function also without clinical heart failure and independent of traditional cardiovascular risk factors, for example, hypertension and diabetes.23 This provides further links between systemic inflammation and myocardial disease, which could sensitise to overt heart failure in the advent of additional stress factors, for example, hypertension or ischaemia.
Structural and functional defects of the Ca2+ handling system represent an important feature of cardiac hypertrophy, heart failure or ventricular arrhythmia.3 Mechanisms implicated in these abnormalities include RyR2 phosphorylation and redox modification of RyR2.16 Our data show that cardiomyocytes isolated from CAIA animals display reduced global [Ca2+]i transients and SR Ca2+ stores. This can be seen in the context of increased oxidative modifications of RyR2 in hearts of CAIA mice. RyR oxidation, in various conditions including ageing or heart failure, can lead to diastolic Ca2+ leak that in turn is linked to reduced SR Ca2+ stores.8 16 Moreover, we found calsequestrin proteins to be oxidatively modified. Functional consequences of oxidative modifications of calsequestrin have, to our knowledge, not been extensively studied. Yet, it is conceivable that oxidation of calsequestrin can impact SR Ca2+ buffering, which is seen with other calsequestrin modifications.24
The L-type Ca2+ current is a major determinant of the cardiomyocyte action potential and has a critical role for excitation–contraction coupling in the heart. CAIA myocytes display reduced L-type Ca2+ currents that would dampen the global [Ca2+]i transient (figure 4). The effects of oxidation-dependent modifications in the L-type Ca2+ channel complex are not completely understood. However, there are evidence that exogenous oxidative agents can reduce the L-type Ca2+ current. Other studies have shown that ROS may even potentiate the L-type Ca2+ current indicating that the L-type Ca2+ currents may be affected by redox-dependent mediators in a biphasic manner, for example, as a function of the magnitude and duration of the oxidative signal. Taken together, our data show that hearts of CAIA mice display oxidation-dependent modifications of the major Ca2+ handling proteins (RyR2, DHPR and calsequestrin) that is linked to impaired cardiomyocyte Ca2+ handling.
It is likely that factors released during chronic inflammation, such as cytokines, play a central role in mediating RA-dependent cardiac remodelling. Interestingly, blocking interleukin 1β (IL-1β) has been shown to improve left ventricular function in patients with RA25 and in clinical cases to resolve refractory constrictive pericarditis.26 Furthermore, IL-1β-lowering therapy reduces morbidity in clinical studies on cardiovascular disease.27 28 Surprisingly, the CAIA mice showed no significant increase in the concentration of IL-1β (online supplementary figure S1C). This could be due to that IL-1β increases at a time point outside of what we have measured in this study. However, blocking Il-1β can improve cardiovascular outcome also without detectable IL-1β in serum.28 In contrast, the cytokine TNF is robustly upregulated both in the hearts and serum of CAIA mice and in patients with RA.12 Furthermore, TNF can stimulate ROS production in cardiomyocytes6 and promote cardiomyopathy and heart failure.21 We cannot exclude that other factors are also involved in the pathogenic signalling. Interestingly, patients with RA display increased levels of microparticles (eg, exosomes) in plasma29 that could play a role in cardiovascular disease.30 Our findings are consistent with a model whereby inflammatory factors released during the chronic inflammatory disease mediates myocardial remodelling (figure 6).
In conclusion, our study shows molecular and functional maladaptations to the heart in the context of RA. This includes: (1) myocardial remodelling with hypertrophy and increased fibrotic deposition, (2) impaired cardiomyocyte Ca2+ signalling, (3) mildly reduced cardiac contractility and (4) oxidation-dependent modification of important Ca2+ handling proteins. Together, our data show that inflammatory arthritis induces long-lasting heart remodelling that are linked to impaired cardiac function. This is consistent with an acquired non-ischaemic cardiomyopathy that, translated to patients, may represent subclinical heart disease in RA. The presence of cardiomyopathy in this model highlights an important comorbidity in inflammatory rheumatic disease that has implications to screening and preventive cardiac management in patients with RA.
Key messages
What is already known on this subject?
Rheumatoid arthritis (RA) can affect the heart leading to cardiovascular disease that is a major comorbidity and can remain clinically silent without detailed cardiovascular investigation.
What might this study add?
This study shows for the first time in a mouse model of RA an acquired form of cardiomyopathy with hypertrophy, fibrosis signalling, oxidative stress and impaired contractile function.
How might this impact on clinical practice?
The risk of developing heart failure in RA is significantly elevated both in the active inflammatory state and during remission. Yet, the molecular changes that occur in the heart during RA is little understood. This study shows that cardiac molecular and physiological changes are present also when the arthritis activity is in remission. This underscores the importance of following patients with RA for development of heart failure.
Acknowledgments
We would like to thank Professors Anders Arner and Bo Rydqvist for technical assistance on echocardiography and patch clamp experiments, respectively.
References
Footnotes
Contributors GP contributed to study conception and design, acquisition of the data, analysis and interpretation of the data and drafting the article. DCA contributed to study conception, analysis and interpretation of the data and writing the article. CIS contributed in acquisition and interpretation of the data. LHL contributed to analysing and interpreting the data. AB-F, NMA, KS, TF-Z and AJ contributed in acquiring and analysing the data. All the authors contributed in revising the article critically for important intellectual content. All the authors approved the final version of the article to be published.
Funding This work was supported by the Lars Hiertas foundation to GP (FO20150396), the Swedish Heart Lung Foundation (20160741 and 20150651), the Stockholm County Council (20120687), Åke Wiberg Foundation, The Jeansson Foundation and The Swedish Society for Medical Research to DCA, the Swedish Research Council (542-2013-8373), Knut and Alice Wallenberg Foundation, Ragnar Söderberg Foundation and EU Project FP7-Health-2013-Innovation (1602919-2) to CIS, the Swedish Heart Lung Foundation (20150557) and Swedish Research Council grants (523-2014-2336 and 2013-23897-104604-23) to LHL.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.