Article Text

Abstract
Late sodium current in cardiac cells is very small compared with the fast component, but as it flows throughout the action potential it may make a substantial contribution to sodium loading during each cardiac cycle. Late sodium current may contribute to triggering arrhythmia in two ways: by causing repolarisation failure (early afterdepolarisations); and by triggering late afterdepolarisations attributable to calcium oscillations in sodium–calcium overload conditions. Reduction of late sodium current would therefore be expected to have therapeutic benefits, particularly in disease states such as ischaemia in which sodium–calcium overload is a major feature.
- EAD, early afterdepolarisation
- IbNa, sodium background current
- ICa, L, L-type inward calcium current
- IKr, rapid delayed potassium current
- IpNa, persistent sodium current
- [Na+]i, intracellular sodium concentration
Statistics from Altmetric.com
- EAD, early afterdepolarisation
- IbNa, sodium background current
- ICa, L, L-type inward calcium current
- IKr, rapid delayed potassium current
- IpNa, persistent sodium current
- [Na+]i, intracellular sodium concentration
The upstroke of the cardiac ventricular action potential is generated by activation of tetrodotoxin-sensitive sodium channels. The rapidity of the depolarisation is responsible for the fast activation wave front ensuring that the whole ventricle is excited within around 20 ms, so enabling nearly synchronous initiation of contraction. The sodium channel inactivation process is also rapid. It would be wasteful for a high level of sodium entry to continue throughout the action potential plateau. The inactivation process is not complete, however. The earliest models of the action potential based on the Hodgkin–Huxley equations for the sodium current predicted that around 1–2% of the peak current would be expected to flow during the relatively low plateau of Purkinje fibres,1 so enabling reconstruction of Weidmann’s experiments showing action potential shortening when external sodium is reduced.2 This effect arises from a small degree of overlap between the activation and inactivation curves, so that there is a window of potentials over which a fraction of sodium channels are activated but not inactivated. This is known as the sodium window current.3
Although the predicted sodium window can account for some of the late sodium current, it cannot account for all of it. Patch-clamp recordings of single sodium channels show late openings of channels at voltages outside the predicted window, and this is confirmed by whole-cell recordings.4 In particular, late sodium current flows at positive membrane potentials where the Hodgkin–Huxley equations in the cardiac cell models would predict virtually zero current. This is important, as the ventricular action potential plateau in species such as human, dog and guinea pig occurs mostly at positive membrane potentials. There is therefore a continued inward flow of sodium current throughout the plateau in ventricular cells as well as in Purkinje fibres. Sodium loading during the action potential therefore occurs in two phases: an intense phase lasting only a few milliseconds and a weak phase lasting hundreds of milliseconds. Even though the weak phase is a tiny trickle—around 1–2% compared with the peak current—it lasts for around 50–100 times longer, so the net sodium loading by the two phases during the complete cardiac cycle is comparable. Reducing the late sodium current could therefore reduce sodium loading by up to around 50%.
Another feature that distinguishes much of the late sodium current from the predicted window current is that, at a constant voltage, it slowly inactivates, whereas the window current would be expected to be a constant once the fast inactivation phase has reached a steady state.
The first experimental indications for slowly inactivating, persistent sodium current, now called IpNa, came from studies on Purkinje fibres of dogs and rabbits.5,6 This was followed by the discovery of IpNa in ventricular myocytes of rats.7 Subsequently, IpNa was described in guinea pig ventricular myocytes by using single-channel and whole-cell patch clamping.8 This study also suggested a significant effect of IpNa on action potential duration in ventricular cells: application of 60 mmol/l tetrodotoxin reversibly shortened action potential duration at 95% repolarisation by about 10%. Computer modelling studies also suggest that the magnitude of IpNa in guinea pig ventricular myocytes can have significant effects on the duration of the action potential plateau.4,9
The role of modulation of IpNa in disease was highlighted by studies investigating the changes in IpNa in rat ventricular myocytes in the absence and presence of hypoxia.10,11 IpNa increased during hypoxia, and it was suggested that it could be involved in the development of early afterdepolarisations (EADs) and arrhythmias during hypoxic states. Maltsev et al12 showed that IpNa is present in ventricular myocytes from human mid-myocardium in normal donor hearts and patients with heart failure. They found a similar (15–20%) reduction in action potential duration after application of 1.5 mmol/l tetrodotoxin. They also showed that tetrodotoxin abolished EADs in myocytes isolated from patients with heart failure. Computer modelling studies reproduce these data well.13 It has been shown that EADs can be induced in a cell model by increasing IpNa. One mechanism for this is that the inactivation curve of the fast sodium current is shifted in the depolarising direction, as has been observed in an SCN5A missense mutation.14 An increase in late sodium current is also present in some well-known genetic predispositions to arrhythmias—for example, one type of the long-QT syndrome.15
IpNa is distinct from the sodium background current (IbNa), which is tetrodotoxin insensitive and shows no voltage dependence.16 In an integrative analysis of sodium loading it is important to take account of IbNa. Like IpNa, it is a small current but because it flows continuously it contributes to sodium homeostasis. The influence of this sodium “leak” current was analysed by Eisner,17 who showed its importance in determining the relative magnitude of frequency changes on sodium loading and on contraction. As yet, the molecular and genetic origins of IbNa are unknown. There have been speculations that it may be a leak form of sodium–potassium ATPase (sodium pump)18 or of sodium–calcium exchange.19
METHODS
The computations described in this article were done with OXSOFT HEART V.4.8 and V.4.x (Oxsoft, Oxford, UK) on previously described models.4,9,20–22
Distribution of IpNa within the ventricle
Ion channels and transporters are not uniformly distributed throughout the ventricle. Heterogeneities of expression or regulation underlie the well-known differences in action potential shape and duration, as between apex and base and across the ventricular wall. These differences are responsible for the direction and form of the T wave of the ECG,23 and they have been modelled in some detail.24
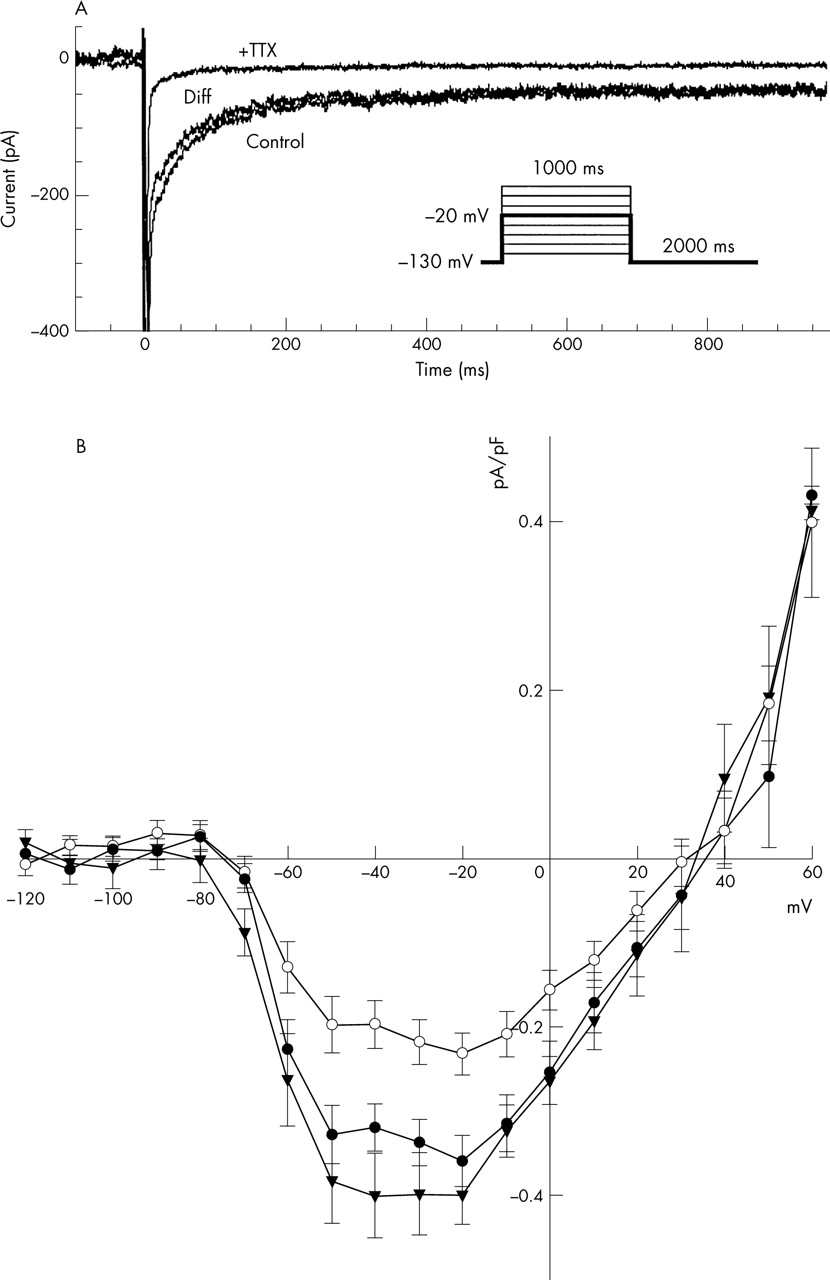
Work in our laboratory on cells isolated from different depths within the ventricular wall found that in guinea pig, IpNa is smallest in the mid-myocardial cells (fig 1). This result was surprising, as it has been assumed to be largest in mid-myocardial cells where the action potential is longest and where cells characterised as M cells have been found.25 This difference can be explained in several ways. IpNa could be smallest in M cells despite their long action potential duration, assuming that differences in other currents, such as IK, are responsible for their long action potential duration. Alternatively, M cells could be a small subset of all mid-myocardial cells and, therefore, only occasionally observed in mid-myocardial single-cell preparations. In agreement with this, several studies failed to find significant evidence of M cells in guinea pig, rat and pig hearts.26–28

(Top) Current recordings obtained during depolarising voltage-clamp pulses from −130 to −20 mV (see inset). The traces show recordings during control conditions (control) and after application of 63 μmol/l tetrodotoxin (+TTX) and the difference current (Diff). (Bottom) Current–voltage relations of the persistent sodium current (IpNa) in guinea pig ventricular myocytes from the subendocardium (•), mid-myocardium (○) and subepicardium (▾). Data points were measured as current density averages between 300–350 ms after the onset of depolarisation. Note that IpNa flows at all potentials between the threshold (about −70 mV) and the reversal potential (about 40 mV). Current flow is therefore significant at plateau potentials, typically between 20 and −20 V.4
The existence of IpNa is one of the factors predisposing cardiac cells to failure of repolarisation or the initiation of EADs and, possibly, re-entrant arrhythmias. Any inward current flowing during the late plateau phase will have these effects, and they are produced by drugs that reduce the rapid delayed rectifier current (IKr) (HERG-acting drugs) and by certain genetic mutations.29 It is already known, however, that combining HERG-blocking action with block of the L-type inward calcium current (ICa,L) protects against repolarisation failure.30 We may therefore expect a similar protection when HERG block is combined with block of IpNa. Figure 2 shows this effect in a guinea pig ventricular cell model modified to include IpNa to fit the results of Sakmann et al.4
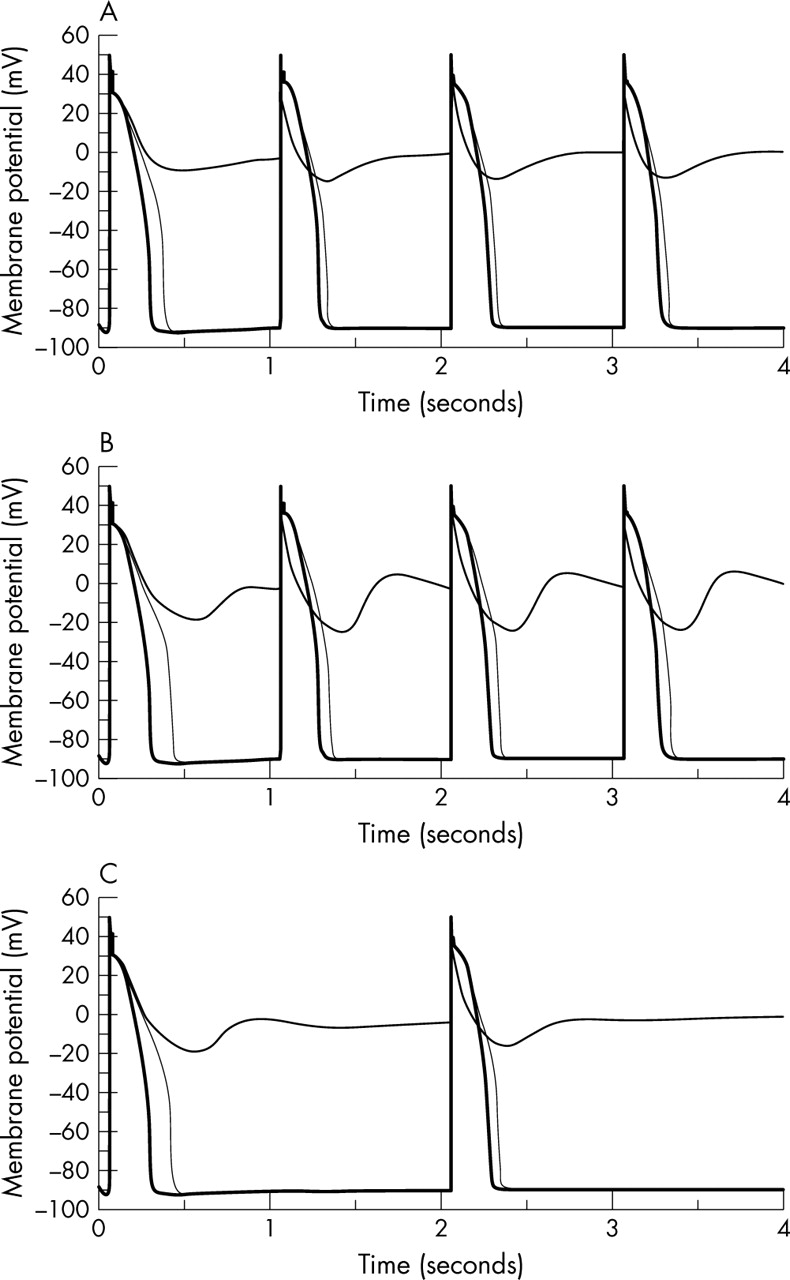
Simulated membrane potential traces from the Sakmann model4 based on Noble et al.9 (Top) Control (thick line), 100% block of the rapid delayed rectifier current (IKr) (middle line) and 100% IKr + 100% persistent sodium current (IpNa) block (thin line) at 3.5 mM external potassium concentration ([K+]o) and 1 Hz. (Middle) Control (thick line), 90% IKr block (middle line) and 90% IKr + 50% IpNa block (thin line) at 3.5 mM [K+]o and 1 Hz. (Bottom) Control (thick line), 90% IKr block (middle line) and 90% IKr + 50% IpNa block (thin line) at 3.5 mM [K+]o and 0.5 Hz. Computations were done specifically for this article.
In this model we can induce repolarisation failure by blocking IKr at an external potassium concentration of 3.5 mmol/l. Additional block of IpNa then restores smooth repolarisation. A compound that has this combination of actions should not therefore be proarrhythmic through repolarisation failure. It is important to note, however, that we would expect this potentially beneficial action profile to hold good only for compounds that act on IpNa without significantly affecting the peak sodium current on which rapid conduction depends. Blockers of the fast sodium current are proarrhythmic because they slow conduction and so allow re-entry to develop more readily.
Sodium loading during repetitive activity
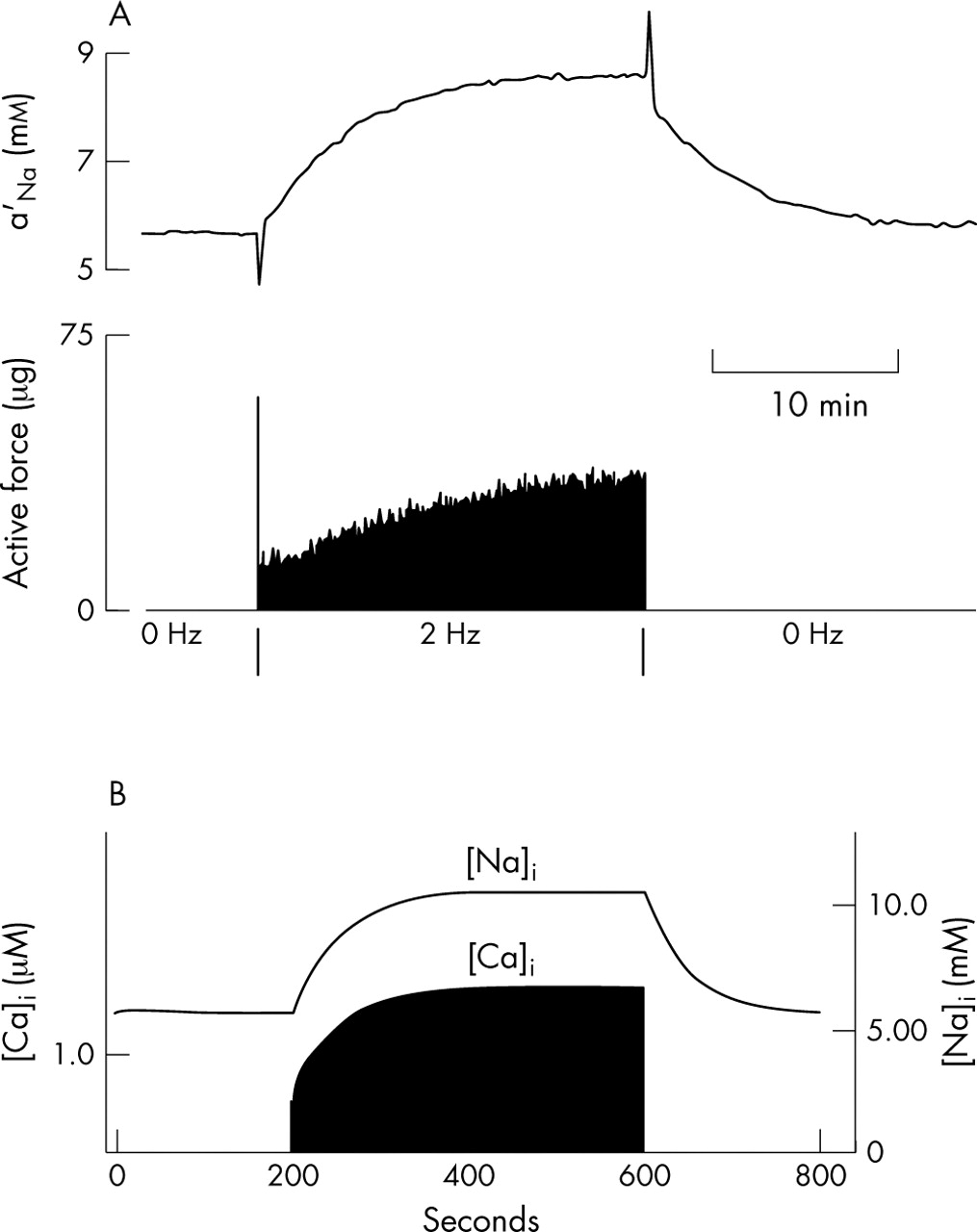
In a resting cardiac cell the inward sodium leak must be balanced by outward sodium pumping mediated by sodium–potassium ATPase (the sodium pump). During activity, extra sodium enters cells through sodium channels and through sodium–calcium exchange. Intracellular sodium concentration ([Na+]i) therefore rises until a new balance is achieved when activation of the sodium pump by increased internal sodium becomes sufficient to cope with the extra load. Figure 3 (top) shows an experiment in which intracellular sodium activity and force of contraction were recorded simultaneously in a cardiac Purkinje fibre during rest and during a period of repetitive activity at a frequency of 2 Hz, similar to that during normal beating of the heart.31,32 The resting concentration of internal sodium activity was just over 5 mmol/l. During repetitive activity it rose over a period of several minutes to a new steady state at just under 9 mmol/l, corresponding to nearly a doubling of [Na+]i. Force of contraction also rose progressively, an effect that is known to be a secondary consequence of a rise in [Na+]i.17 The lower part of this figure shows that the effect is well reproduced by models of the ventricular action potential. The model used here was developed for guinea pig ventricular cells.9 The magnitude and speed of rise in [Na+]i are very well reproduced. The model gives rather higher values, with a resting concentration a little more than 5 mmol/l and a concentration during activity a little more than 10 mmol/l. The doubling of sodium concentration is therefore well reproduced.

(Top) Intracellular sodium activity and tension recorded in a cardiac Purkinje fibre during rest and during a period of repetitive stimulation at 2 Hz.31 (Bottom) Reconstruction of a model ventricular cell9 showing intracellular sodium ([Na]i) and calcium ([Ca]i) concentrations. The computations for this figure were done specifically for this article.
The relative magnitude of the rise in tension is well reproduced by the calcium signal in the model (lower trace), but time course differs significantly. The model calcium concentration closely follows the time course of rise in [Na+]i, whereas the rise in recorded tension in the experiment is significantly delayed and then continues even when the sodium activity is nearly constant. This difference may be attributable to the observed hysteresis in force development during rises and falls of [Na+]i.33 The mechanism for this, which is still unclear, is not represented in the model.
Late afterdepolarisations caused by sodium–calcium overload
EADs are an immediate consequence of increased sodium entry during action potentials, as they arise from IpNa interrupting repolarisation, perhaps in combination with disease, drug or genetic factors. This effect can occur even during a single cardiac cycle. Longer-term consequences of sodium loading also arise from increased sodium entry during electrical activity.34 The sequence of events is now well understood. Sodium loading leads, through sodium–calcium exchange, to calcium loading. The sodium gradient becomes insufficient to maintain the calcium gradient, so intracellular calcium increases. Up to a certain level, this is a well-behaved process: initially the change in calcium is a monotonic function of the change in sodium. But beyond a threshold, which depends on many factors but is typically found when [Na+]i exceeds around 12–15 mmol/l (that is, three times normal sodium concentrations), the rise in calcium triggers repetitive release of calcium from the sarcoplasmic reticulum. This process is a pathological version of the calcium-induced calcium release process first identified by Fabiato,35 which is part of the normal process of excitation–contraction coupling.36
Figure 436,37 illustrates a simulation of this phenomenon in a cardiac cell model in which [Na+]i rose after inhibition of the sodium–potassium pump.34 In this example, oscillatory calcium release begins at [Na+]i around 14 mmol/l. Similar results have been obtained in simulations of ischaemic conditions.36
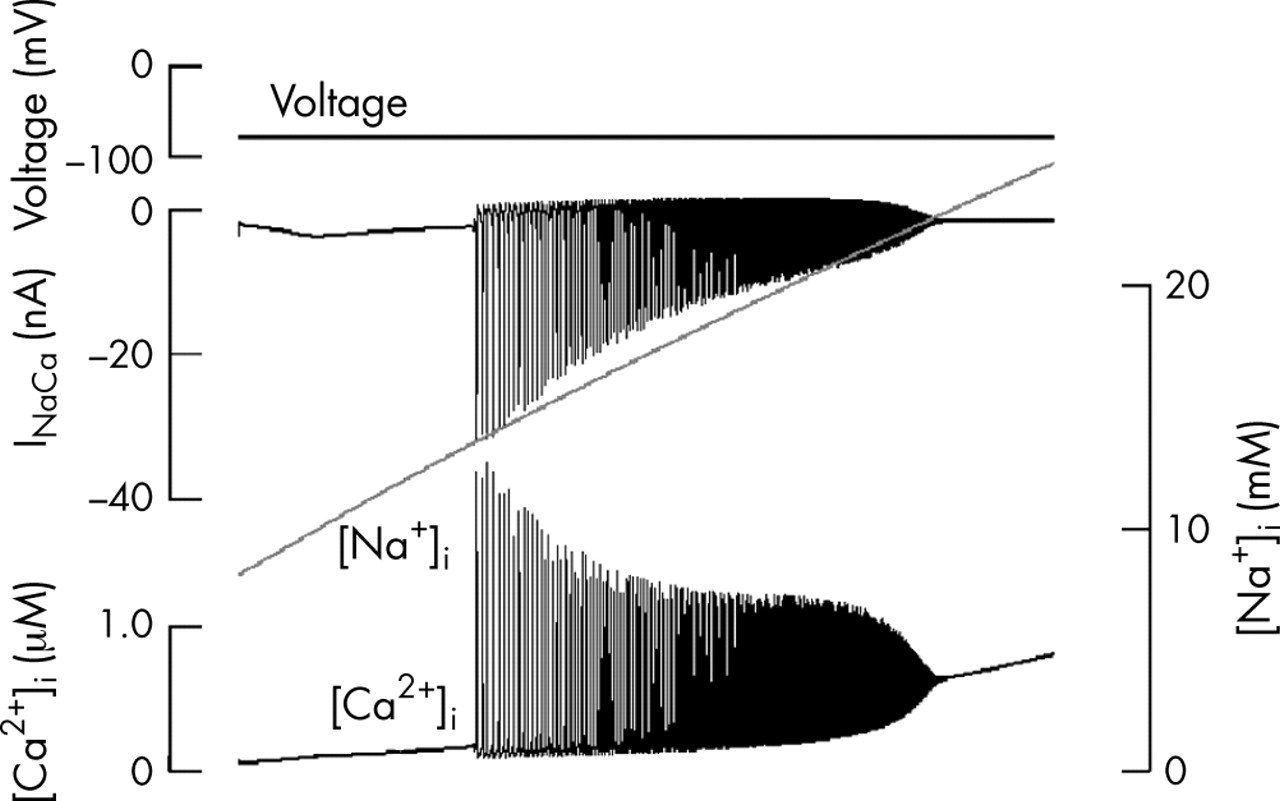
Reconstruction of the events during sodium–calcium overload resulting from inhibition of the sodium pump. This computation uses a partial model of ischaemia.36 It isolates the part of the process that is secondary to the rise in intracellular sodium concentration ([Na+]i). Records from top downwards: membrane voltage (held constant in this computation), sodium–calcium exchange current (INaCa), [Na+]i and intracellular calcium concentration ([Ca2+]i). The model is based on a multicellular Purkinje fibre model,20 which is why the exchanger current is plotted in nA rather than pA, as would be appropriate in a single cell.37
The mechanism by which oscillatory calcium release can generate arrhythmias is also understood. Each oscillation of calcium activates the sodium–calcium exchange, which is electrogenic. As the sodium gradient is used to pump calcium out of the cell, the exchanger carries an inward current. If this current is sufficiently large, it can trigger additional action potentials and so form a mechanism for arrhythmias based on ectopic beating. Figure 5 shows this phenomenon in a model of atrial cells.21,22

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanism by which ectopic beating may be triggered during sodium–calcium overload. In these computations intracellular sodium concentration ([Na+]i) was set at 16.24 mmol/l, corresponding to [Na+]i reached during ischaemia (compare fig 4). A single stimulus was then applied to initiate the first action potential in the series (top trace). The subsequent action potentials are spontaneous. They arise from calcium oscillations (middle trace) similar to those illustrated in fig 4. The bottom trace shows the computed sodium–calcium exchange current. Each rise of intracellular calcium concentration ([Ca2+]i) generates an inward (downward) exchange current, which rapidly reverses during the action potential upstroke but then becomes inward again during the late plateau. The initial inward surge is responsible for triggering each ectopic excitation.34
DISCUSSION
Mechanisms of arrhythmia and logic of sodium channel drugs
There are at least two mechanisms by which raised late sodium current can trigger arrhythmias. The first is immediate and is dependent on interruption of repolarisation, leading to EADs and the initiation of torsade de pointes arrhythmia. The second is more gradual in onset and is attributable to intracellular sodium loading, leading to calcium overload, oscillatory calcium release and the initiation of late (delayed) afterdepolarisations generated by the sodium–calcium exchange current activated by each rise in intracellular calcium. Both of these mechanisms are now sufficiently well understood to be simulated successfully in computer models of cardiac cells.
In both mechanisms, drugs that reduce late sodium current would be expected to have therapeutic potential by reducing or eliminating the probability of occurrence of EADs and delayed afterdepolarisations. The important qualification to be noted is that the action should necessarily be limited to the late component of sodium current. Reduction of the peak sodium current would itself promote arrhythmia by slowing conduction and enabling re-entry to occur more readily.
The discovery of a compound that reduces IpNa without significant action on peak sodium current at therapeutic levels is therefore an exciting and promising development.38
Acknowledgments
Work in our laboratory is supported by grants from the EU BioSim Consortium, the Wellcome Trust, the EPSRC and the MRC.
REFERENCES
Footnotes
-
Competing interests: None declared.