Article Text

Abstract
The structural and functional abnormalities that lead to cardiac death are coronary artery disease and left ventricular abnormalities related to remodelling (left ventricular hypertrophy, left ventricular systolic dysfunction, and left ventricular fibrosis). Aldosterone adversely affects all of these processes. It produces both a vasculopathy and left ventricular dysfunction and fibrosis. Endothelial dysfunction in the coronary arteries can lead to acute coronary events. Left ventricular dysfunction will cause the progression of heart failure, and left ventricular fibrosis and dysfunction provide an arrhythmic substrate. The combination of acute coronary events and arrhythmias can lead to sudden cardiac deaths, while acceleration of the heart failure disease process can lead to deaths from progressive heart failure. The increased understanding of the mechanistic role of aldosterone in cardiovascular disease provides a rationale for the positive results that have been seen in clinical trials of aldosterone blockade.
- ACE, angiotensin converting enzyme
- COX-2, cyclo-oxygenase- 2
- L-NMMA, N-monomethyl-L-arginine
- MI, myocardial infarction
- PIII NP, procollagen type III amino terminal peptide
- VCAM-1, vascular cell adhesion molecule-1
- aldosterone
- heart failure
- myocardial infarction
Statistics from Altmetric.com
- ACE, angiotensin converting enzyme
- COX-2, cyclo-oxygenase- 2
- L-NMMA, N-monomethyl-L-arginine
- MI, myocardial infarction
- PIII NP, procollagen type III amino terminal peptide
- VCAM-1, vascular cell adhesion molecule-1

VASCULAR EFFECTS OF ALDOSTERONE
Aldosterone produces a vasculopathy that appears to affect the coronary arteries. Endothelial function has been measured in the brachial artery of patients with heart failure, with the response to acetylcholine being taken as a measure of the health of the endothelium.1 In patients given aldosterone blockade there was an approximate doubling in the forearm blood flow response to acetylcholine. Aldosterone blockade also increased the vascular response to N-monomethyl-L-arginine (L-NMMA).
A study in patients with mild heart failure who were already taking a β blocker, statin, and angiotensin converting enzyme (ACE) inhibitor has also demonstrated that in the presence of aldosterone blockade there was an improvement in acetylcholine mediated, endothelium dependent vasodilation.2 It has also been shown that infusion of aldosterone for one hour in healthy individuals (in a dose that does not change the haemodynamics) leads to development of endothelial dysfunction, identified as a notably reduced vascular response to acetylcholine.3
Heymes and colleagues have reported research with transgenic mice that over express aldosterone synthase in the myocardium. They found that the transgenic animals had normal cardiac function but major coronary artery dysfunction, which for some reason was more pronounced in males.4
The human and transgenic animal data therefore both indicate that aldosterone produces endothelial dysfunction. The mechanism is thought to involve reduction in vascular nitric oxide by increased production of superoxide anions. Superoxide anions degrade the nitric oxide that is produced by healthy endothelium. Experiments in hyperlipidaemic rabbits have shown a 2.3-fold increase in superoxide anion generation that was reversed when the animals were given aldosterone blockade.5
Animal studies have suggested an inflammatory component to aldosterone induced vasculopathy. Administration of aldosterone to rats has been shown to be associated with macrophage and monocyte infiltration (a measure of inflammation) within and around the coronary artery wall.6 Other inflammatory signals were also demonstrated, with both vascular cell adhesion molecule-1 (VCAM-1) and cyclo-oxygenase-2 (COX-2) being produced in the coronary arteries.6
All of this work suggests that a vasculopathy of some kind is produced by aldosterone and, at least in animals, this appears to have an inflammatory component.
EFFECT OF ALDOSTERONE ON ANGIOTENSIN
Another vascular effect of aldosterone is that it appears to be able to feedback on the rest of the renin–angiotensin system and to amplify the production of angiotensin II from angiotensin I (fig 1). If this positive feedback loop is true, it suggests that administration of an aldosterone blocking drug will not only block the effects of aldosterone but as a “byproduct” it will also produce some additional ACE inhibition, by blocking the conversion of angiotensin I to angiotensin II.

Aldosterone is thought to amplify production of angiotensin II from angiotensin I and hence aldosterone blockade produces some “extra” ACE inhibition.
This is something of a pharmacological curiosity at present but there are some supporting data. In the early 1990s, Sun et al7 showed in animal experiments that aldosterone significantly increased the binding density of ACE receptors, while Ullian et al8 showed that aldosterone increases angiotensin II receptor number. More recently, researchers in Japan9 have shown that the addition of aldosterone to cultured rat cardiomyocytes led to a dramatic 23-fold increase in production of ACE messenger RNA. The effect of aldosterone was time and dose dependent, and it was significantly inhibited by administration of the aldosterone antagonist spironolactone.
These in vitro studies indicate that some ACE inhibition may be occurring with aldosterone blockers and a clinical study that we carried out showed similar effects.1 Forearm blood flow response to angiotensin I and II was measured after one month’s treatment with spironolactone or placebo. The vascular response to angiotensin I was blunted in the presence of aldosterone blockade, suggesting that less angiotensin I was being converted to angiotensin II, since vasoconstriction only occurs on the conversion to angiotensin II.
There is therefore a developing body of evidence that there is some feedback amplification and that aldosterone blockade, in addition to its other benefits, produces some “extra” ACE inhibition.
ALDOSTERONE AND LEFT VENTRICULAR ABNORMALITIES
Studies investigating the effects of aldosterone on left ventricular abnormalities have shown a consistent pattern. It appears that aldosterone is able to produce tissue injury, including injury to the myocardium, the consequences of which include left ventricular dysfunction and left ventricular fibrosis.
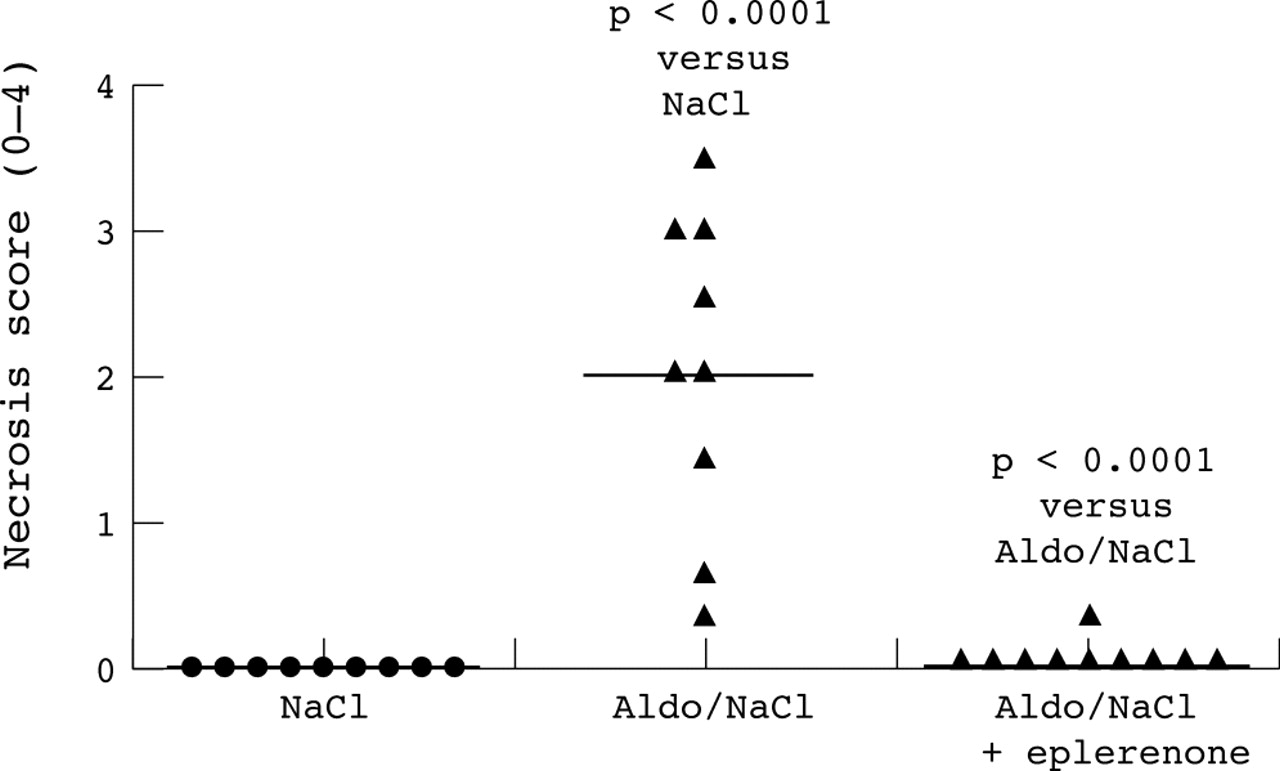
Animal studies have provided evidence that aldosterone blockade can prevent aldosterone induced myocardial injury. In rats with hypertension induced by aldosterone and saline, Rocha et al6 demonstrated that myocardial injury, as measured by necrosis score, was increased by aldosterone and brought back to control level when the animals were given the aldosterone antagonist eplerenone (fig 2). Similar effects have also been seen outside the heart: in spontaneously hypertensive rats, cerebral tissue injury appeared to be notably reduced by administration of eplerenone at a dose that did not affect the blood pressure.10

Aldosterone blockade with eplerenone was shown to prevent aldosterone induced myocardial injury. Adapted with permission from Rocha et al.6
Suzuki and colleagues reported experiments investigating the effect of three months’ treatment with eplerenone on the progression of left ventricular dysfunction and remodelling in dogs with chronic heart failure induced by intracoronary microembolisations that were discontinued when left ventricular ejection fraction was between 30–40%.11 The difference between the eplerenone group and a control group in left ventricular end diastolic pressure, left ventricular end diastolic volume, and end diastolic wall stress—parameters of left ventricular remodelling—were all significant and favoured eplerenone, indicating that aldosterone blockade can reduce myocardial remodelling and reduce wall stress after induction of heart failure.
In a clinical study in patients with mild heart failure,2 left ventricular mass index (a measure of hypertrophy) and left ventricular internal diastolic diameter were both improved by aldosterone blockade with spironolactone, providing further evidence that aldosterone blockade reduces left ventricular remodelling (fig 3). An Italian study has produced similar findings, with reported improvement in both left ventricular ejection fraction and maximum oxygen consumption in patients with chronic heart failure given aldosterone blockade with spironolactone.12

In patients with heart failure, left ventricular mass index and left ventricular internal diastolic diameter were both improved by aldosterone blockade, indicating that treatment can reduce left ventricular remodelling (MacDonald et al2).
In addition to left ventricular dysfunction, left ventricular fibrosis is a key characteristic of aldosterone induced tissue injury. Eplerenone has been shown to prevent vascular injury in the myocardium of rats with hypertension induced by aldosterone and saline. Lesions in the myocardial tissue of these rats were primarily localised within and around coronary blood vessels, and were characterised by intense infiltration of inflammatory cells. These fibrotic lesions were absent following administration of eplerenone to the aldosterone/saline treated rats.
Clinically, the effect of aldosterone blockade on myocardial fibrosis can be demonstrated by measuring collagen markers in the blood stream. Procollagen type III amino terminal peptide (PIII NP) is a marker that can be taken as an index of myocardial collagen turnover. There is a considerable amount of data showing a significant reduction in PIII NP blood values in patients with chronic heart failure who are taking the aldosterone blocker spironolactone.13
Chronic heart failure and post-myocardial infarction (MI) heart failure are very similar entities, but there are also some data on the post-MI situation per se. Measuring endothelial function in patients early after MI is difficult but animal studies have been carried out. In one study, vascular response to acetylcholine was measured after MI in animals given either eplerenone, the ACE inhibitor trandolopril, or a combination of the two drugs. Animals that received the combination had the best endothelial function, which was similar to that in control animals.14 It was also shown that superoxide anion production in these post-MI animals was notably increased, but production was reduced by aldosterone blockade, whether or not trandolopril was also given.
A clinical study reported from Japan15 investigated left ventricular function in post-MI patients given spironolactone. Indices of left ventricular remodelling, such as left ventricular ejection fraction, end diastolic volume index, and left ventricular end systolic volume index, were all notably improved in patients who were given spironolactone (by 4–18%). Reduction in the collagen marker (PIII NP) was also shown, and there was a correlation between the change in post-MI end diastolic volume index and the change in blood concentrations of the collagen marker. This suggests that left ventricular dysfunction and left ventricular fibrosis are parallel processes, both of which are favourably influenced by aldosterone blockade.
The data discussed above indicate that aldosterone is involved in promoting a vasculopathy in the arteries, including the coronary arteries, and that it is also involved in the post-MI situation in producing left ventricular dysfunction and fibrosis. The possible sequence of events and the probable clinical consequences are shown in fig 4. Our current understanding of events is that endothelial dysfunction in coronary arteries is likely to lead to development of acute coronary events. Left ventricular fibrosis and dysfunction are thought to be arrhythmogenic stimuli and the combination of acute coronary events and an arrhythmogenic tendency are likely to increase the risk of sudden cardiac death. In addition, progressive decline in left ventricular systolic function and hence acceleration of the heart failure disease process can lead to earlier death from progressive heart failure.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Suggested scheme for the mechanistic role of aldosterone in cardiovascular disease.
Alongside the increased understanding of the mechanistic role of aldosterone in cardiovascular disease, there is now evidence from two clinical trials16,17 showing that aldosterone blockade can indeed reduce both sudden death and the progression of heart failure.
Learning points
-
Aldosterone adversely affects both vascular and myocardial function
-
There is evidence to suggest that aldosterone produces coronary endothelial dysfunction and left ventricular dysfunction and fibrosis
-
Because of the adverse effects of aldosterone on both blood vessels and the heart, aldosterone blockade reduces both sudden cardiac deaths and the progression of heart failure
CONCLUSION
There is now an increased understanding of the mechanistic role of aldosterone in cardiovascular disease. Aldosterone appears to be able to produce endothelial dysfunction, which may increase the risk of acute coronary events. It also produces left ventricular dysfunction and fibrosis. The reduction in cardiac deaths that has been observed with aldosterone antagonist treatment in patients with heart failure is probably related to both reduction in new coronary events and reduction in left ventricular dysfunction and fibrosis.