Article Text

Abstract
Treatment of children and adults with pulmonary hypertension (PH) with or without cardiac dysfunction has improved in the last two decades. The so-called pulmonary arterial hypertension (PAH)-specific medications currently approved for therapy of adults with PAH target three major pathways (endothelin, nitric oxide, prostacyclin). Moreover, some PH centres may use off-label drugs for compassionate use. Pulmonary hypertensive vascular disease (PHVD) in children is complex, and selection of appropriate therapies remains difficult. In addition, paediatric PAH/PHVD therapy is vastly based on experience and trial data from adult rather than paediatric studies; however, the first randomised paediatric PAH trials have been conducted recently. We present consensus recommendations for the treatment of children with PH. Class of recommendation and level of evidence were assigned based on paediatric data only or on adult studies that included >10% children. After a systematic literature search and analysis of the published data, we developed treatment strategies and algorithms that can guide goal-oriented PH therapy. We discuss early combination therapy (double, triple) in patients with PAH in functional class II–IV and in those with inadequate response to the initial pharmacotherapy. In those children with progressive, severe PAH and inadequate response, advances in drug development, and interventional and surgical approaches provide promising new strategies to avoid, reverse or ameliorate right heart failure and left ventricular compression. In particular, first follow-up data indicate that Potts shunt (left pulmonary artery to descending aorta anastomosis) may be an alternative destination therapy, or bridge to bilateral lung transplantation, in end-stage paediatric PAH.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Pulmonary arterial hypertension (PAH) is still an important cause of morbidity and mortality in children. Despite recent developments in PAH-specific therapies, survival of patients with idiopathic PAH remains poor and appears to be worse in children compared with adults.1 During the past few years, treatment of PAH has undergone a remarkable evolution, which has led to the current approval by regulatory agencies of 10 drugs for adult patients from three main pharmacological groups (addressing three pathways) and four different routes of administration (oral, inhaled, subcutaneous and intravenous). Additional drugs are expected in the near future. Modern drug therapy improves the symptoms of PAH patients and slows down the rates of clinical deterioration. However, emerging therapeutic strategies for adult PAH, such as upfront oral combination therapy, have not been sufficiently studied in children. Moreover, the complexity of pulmonary hypertensive vascular disease (PHVD) in children makes the selection of appropriate therapies a great challenge far away from a mere prescription of drugs. Therapy of paediatric PH is rather characterised by a complex strategy that includes the evaluation of severity and prognosis of the individual disease, the estimation of efficacy of different drugs, and their interaction and combination, as well as supportive and general measures.
Recently, additional surgical and interventional techniques for palliation of children with severe PAH have been reported. The beneficial effects of these strategies are mainly based on the relief of right ventricle (RV) pressure overload with a subsequent reduction of the interventricular septum shift to the left ventricle (LV), and improvement of systolic and diastolic LV performance.
In this paper, we present a comprehensive overview of the current available drug-based and interventional/surgical treatments in paediatric PH and provide the recommendations of the European Paediatric Pulmonary Vascular Disease Network.
Methods
The recommendations given in table 1 relate to the grading system currently suggested by the European Society of Cardiology (ESC) and the American Heart Association, and were based on paediatric data only, or adult studies enrolling >10% children (class of recommendation, level of evidence). The grading and voting process within the writing group is outlined in the executive summary.2 Computerised searches of the PubMed/MEDLINE bibliographic database were conducted between January 1990 and November 2015. Clinical trials, guidelines and reviews limited to paediatric data were searched using the terms ‘pulmonary hypertension’, ‘heart failure’, ‘pharmacotherapy’, ‘drugs’, ‘randomized controlled trial’, ‘atrial septostomy’ and ‘Potts shunt’, in multiple combinations. The primary focus of this paper is on pharmacotherapy and catheter-interventional/surgical treatment of group 1 PH (ie, PAH) according to the World Symposium PH classification, Nice 2013. Therapy of group 2–5 PH is controversial and requires separate discussions. Special considerations for the treatment of PH in the acute and critical care setting,3 treatment of PAH patients with congenital heart disease (CHD)4 and therapy of infants with PH associated with acute and chronic lung disease (CLD)5 are discussed in separate articles of this special issue.
Recommendations on treatment of children with pulmonary hypertension
Pharmacotherapy
Drug therapy in pulmonary hypertension consists of so-called ‘supportive’ and ‘PAH-specific’ therapy (table 1). Detailed information on dosing, adverse effects and other information can be found in table 2.
Pharmacotherapy for paediatric PH
The overall goal of any PH therapy is the transition of a patient from the ‘higher risk’ to the ‘lower risk’ group6 (figure 1).

Determinants of risk in paediatric pulmonary hypertensive vascular disease. The variables listed distinguish between lower risk and higher risk. The intermediate risk group is broad and not specifically defined. Overall, these determinants have only level of evidence C due to sparse or lacking paediatric data. Healthcare providers may include here pulmonary vascular resistance (PVR)/systemic vascular resistance (SVR) ratio, the 6 min walk distance and the max. oxygen consumption (VO2 max.) obtained during cardiopulmonary exercise testing as risk variables; however, it is unclear where exactly the cut-off values should be set. One must also note that most of these variables have been validated mostly for idiopathic pulmonary arterial hypertension (IPAH) and the cut-off levels used above may not necessarily apply to other forms of pulmonary arterial hypertension (PAH). Furthermore, the use of approved therapies and their influence on the variables should be considered in the evaluation of the risk. Modified from McLaughlin et al.6 BNP, brain natriuretic peptide; CI, cardiac index (syn. Qs); mPAP, mean pulmonary artery pressure, mRAP, mean right atrial pressure; NT-proBNP, N terminal pro BNP; PVRi, PVR index; RA, right atrium; RV, right ventricle; SVRi, SVR index.
‘Supportive’ (‘conventional’) therapy
Therapies typically used for left heart failure have been also used for the treatment of patients with RV failure. Supportive therapy may include oxygen, anticoagulants, diuretics, mineralcorticoid receptor antagonists (spironolactone), digoxin. These measures are applied on an individual basis since the currently available studies provide either none or rather ambiguous/contradictory than valid data on most of these therapies in (adults and) children with PH.
Supplemental oxygen
Children with PH and pulmonary diffusion impairment, for example, infants with CLD/bronchopulmonary dysplasia, and those living with PH at high altitude, usually benefit from overnight or continuous oxygen supplementation to overcome chronic alveolar hypoxia. Long-term nocturnal oxygen supplementation improves symptoms in Eisenmenger patients but has not been shown to increase survival.8 Oxygen therapy is reasonable in patients with hypoxaemic PH who have oxygen saturations <92% and/or paO2 <60 mm Hg. Any oxygen supplementation should result in a sustained rise in oxygen saturation and improvement of symptoms.
Diuretics (loop diuretics, thiazides)
Diuretic agents such as furosemide probably have been overused in patients with PAH. Diuretics should be initiated – if at all – cautiously as patients with PAH are frequently preload dependent to maintain sufficient cardiac output.
Digitalis
There are no sufficient clinical data to recommend or refuse the use of digoxin in adult or paediatric PAH patients with RV dysfunction,9 or overt right heart failure.
Mineralcorticoid receptor antagonists (spironolactone)
Spironolactone has only mild diuretic effects but blocks aldosterone and renin angiotensin aldosterone system signalling. RV remodelling and dysfunction appears to be responsive to MR blockade (spironolactone) in the ARIES-1/-2 trials.10 In a post hoc analysis of the ARIES-1/-2 trials, adult patients with PAH who received ambrisentan plus spironolactone had a better outcome than those who received ambrisentan alone.10 Most recently, both plasma aldosterone and its profibrotic target, galectin-3, were found to be increased in adult PAH and associated with WHO functional class, pointing to their role as potential tandem biomarkers.11
Anticoagulation
A potential benefit of long-term anticoagulation (coumadin, warfarin) has not been studied in paediatric PAH, and its use in adults with PAH currently is the subject of an ongoing debate.12 The role of the new oral anticoagulants (NOACs) in PAH is unknown. Thus, aspirin may be considered an alternative to coumadin or warfarin in paediatric idiopathic pulmonary arterial hypertension (IPAH)/heritable pulmonary arterial hypertension (HPAH), particularly in younger, very active children with an assumed higher bleeding risk because of higher accident rates (table 2). The ESC/ERS guidelines for adult PH (2015) carefully outline that anticoagulants may be considered only in adult PAH patients with IPAH, HPAH and PAH associated with anorexigen use (class of recommendation IIb, level of evidence C).12 The use of oral anticoagulants in Eisenmenger syndrome is controversial because the high incidence of PA thrombosis and stroke has to be balanced against the increased risk of haemorrhage and haemoptysis.13 ,14 Anticoagulation is potentially harmful in children with hereditary haemorrhagic telangiectasia 15 or portopulmonary hypertension.
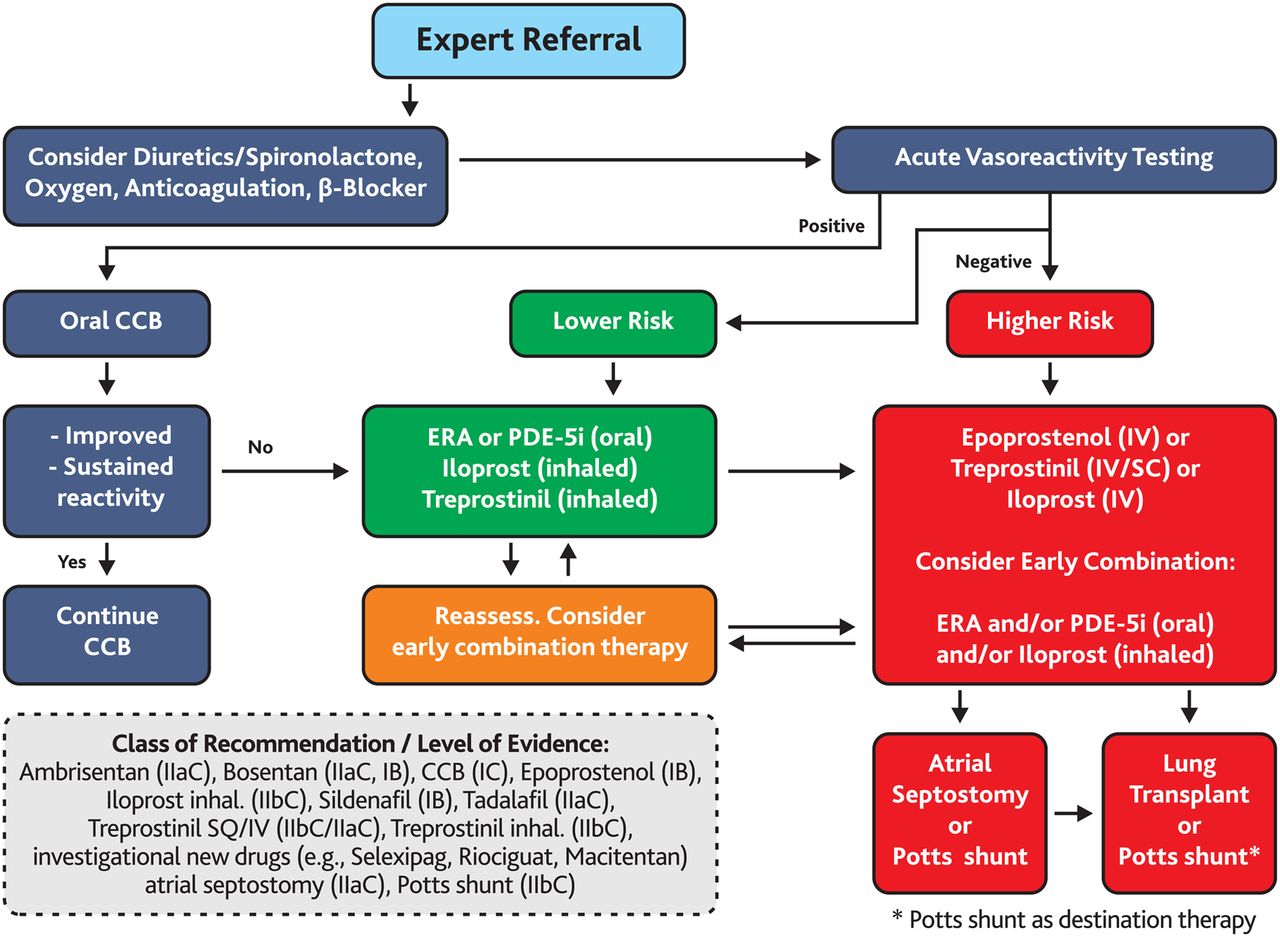
Many PH centres would recommend oral anticoagulation (coumadin, warfarin) in children with high-risk IPAH/HPAH (figures 1 and 2), paediatric PAH patients with low cardiac output, those with hypercoaguable conditions and/or those with long-term indwelling catheters (empiric international normalised ratio (INR) goal 2.0–2.5; some centres recommend INR 1.5–2.0 in young children).

Treatment algorithm for paediatric pulmonary arterial hypertension (PAH). This algorithm applies to idiopathic pulmonary arterial hypertension (IPAH) and hereditary PAH (HPAH (FPAH)). Solid clinical data on the therapy of other forms of PH are limited. The ‘intermediate’ risk group is broad and not specifically defined (see figure 1). Healthcare providers may consider upfront, early or rapid sequence targeted PAH therapy in ‘intermediate’ risk group (between ‘lower risk’ and ‘higher risk’ in figure 1: ‘Determinants of risk’). Use of all agents is considered off-label in children aside from sildenafil and bosentan (>1 year old) in Europe. Sildenafil dosing recommendations should follow the European Medicines Agency-approved dosing for children. Bosentan received the following dual grading: class of recommendation (COR) I, level of evidence (LOE) B for children with PAH and Eisenmenger syndrome (E.S.), and COR IIa, LOE C for children with PAH without Eisenmenger syndrome. Modified and expanded from Ivy et al.7 CCB, calcium channel blocker; ERA, endothelin receptor antagonist; inh., inhalation; IV, intravenous; PDE-5i, phosphodiesterase 5 inhibitor; SC, subcutaneous.
Routine vaccinations are generally recommended for children with PAH: especially infections with influenza, respiratory syncytial virus and pertussis, as well as pneumococcal infections, may lead to PH exacerbation and higher morbidity/mortality.16
For the following discussion of PAH-targeted pharmacotherapies and the according recommendations (tables 1 and 2), we focused the main text on paediatric PAH studies, adult studies that included at least 10% children and the most recent studies (2013–2015) on novel oral compounds (see methods in the executive summary).2 The key outcome data of randomised controlled trials (RCTs) in adult PAH are summarised in tables 3⇓⇓–6.
Characteristics of RCTs with PAH drugs interfering with the endothelin pathway (endothelin receptors antagonists)
Characteristics of RCTs with PAH drugs interfering with the nitric oxide pathway (soluble guanylate cyclase stimulators, phosphodiesterase type-5 inhibitors)
Characteristics of RCTs with PAH drugs interfering with the prostacyclin pathway (prostacyclin analogues and prostacyclin receptors agonists)
Characteristics of RCTs with PAH drugs testing upfront combination therapy
Of note, most PAH-targeted medications are not approved by the European Medicines Agency (EMA) or the US Food and Drug Administration (FDA) for the use in children with PAH (‘off-label’ use).
Calcium channel blockers
Patients who fulfil the criteria of a positive response during acute vasoreactivity testing (AVT) are usually initially treated with an oral calcium channel blocker (CCB) (amlodipine, nifedipine or diltiazem) (table 2). In patients with IPAH/HPAH, the haemodynamic change that defines a positive response to AVT (acute vasoreactivity response (AVR)) in PAH without a shunt (ratio of pulmonary to systemic flow (Qp:Qs)=1:1) for children should be considered as a >20% fall in mean pulmonary artery pressure (PAP) and pulmonary vascular resistance index (PVRi)/systemic vascular resistance index (SVRi) ratio without a decrease in cardiac output (‘AVR in IPAH/HPAH’).17 In the presence of a positive AVR, a fall of the ratio of PVRi/SVRi (or as the authors suggest alternatively, the ratio of the diastolic PAP/diastolic SAP) below 0.4 during the AVT might be an indication for calcium-channel blocker therapy.17 ,18 Of note, the above definition of a positive AVR was designed for the assessment of prognosis and indication for CCB therapy, but not for decision-making on shunt closure in PAH-CHD with elevated PVR.4 ,17
Children with PAH associated with CHD and a significant left-to-right shunt (non-repaired CHD), and those with Eisenmenger syndrome (ie, suprasystemic PVR and right-to-left shunt) most likely do not benefit from CCB therapy.19 It is of great importance to evaluate and reassess the AVT response in children with PAH.17 When the conventional definition of positive AVT (Barst criteria) is applied to the REVAL study, 35% of IPAH/HPAH children but only 15% of those with PAH-CHD were vasoreactive, indicating that children with repaired CHD are less likely to respond during AVT.20 ,21
Children with IPAH who responded to AVT had a survival of 97%, 97% and 81% at 1, 5 and 10 years, respectively; sustained treatment success with CCB was observed, but declined from 84% to 47% in the first 10 years after start of CCB therapy.22 Inadequate use of CCBs in paediatric PAH was associated with higher mortality: children with IPAH who were treated with a CCB regardless of their negative response during AVT had lower survival rates of 45%, 34%, 29% and 29% at 1, 2, 3 and 4 years, respectively.22 ,23
CCBs are contraindicated in non-responders, patients with RV failure (WHO functional class IV)16 regardless of the AVT response, and those who have not undergone AVT.24 The CCBs with significant negative inotropic effects (ie, non-dihydropyridines: verapamil, diltiazem) are usually not recommended for use in the first year of life. However, particularly diltiazem can have additional benefit in children with high heart rates because of its negative chronotropic/dromotropic effects, allowing longer filling and ejection times.
For children with a negative AVT or non-sustained response to CCB, PAH-targeted therapy via oral, inhalative and/or intravenous agents is required (see below). PAH-targeted therapy is currently predominantly based on three major pathways, the NO/cGMP pathway, the endothelin pathway and the prostacyclin pathway.
Endothelin-1 receptor antagonists
Bosentan
Activation of the endothelin system has been demonstrated in patients with PAH, whereas endothelin has vasoconstrictor and mitogenic effects. Bosentan is an oral, dual endothelin A-receptor and B-receptor antagonist (ERA for ETA and ETB). Multiple RCTs on bosentan in adults with PAH (idiopathic, associated with CHD and CTD) showed improvement in exercise capacity, functional class, haemodynamics, echocardiographic variables and time to clinical worsening.
Retrospective observational studies and case series demonstrated that bosentan therapy is safe and appears to be effective in slowing disease progression children with PAH.25 ,26 A retrospective study of 86 children treated with bosentan with or without concomitant therapy was associated with sustained haemodynamic and clinical improvement, and an estimated 2-year survival of 91%.25 However, at 4 years, disease progression in these patients on bosentan was high (54%), with a survival estimate of 82%.27 More than 30 children (≥2 years and <12 years old) were enrolled in the non-randomised FUTURE-1 study (n=36)28 and its extension study, FUTURE-2 (33 children continued on bosentan therapy).29 In the FUTURE-2 study30 (33 children, 2–11.9 years old; median exposure 28 months), adverse events (AEs) that were considered treatment-related occurred in 15 (41.7%) patients. Of 51 serious AEs, 3 were considered treatment-related: two incidences of reported PAH worsening and one of autoimmune hepatitis. Thus, in FUTURE-2, the paediatric bosentan formulation (dispersable tablets; 32 mg) was generally well tolerated, its safety profile comparable to that of the adult formulation when used in children, and the efficacy profile of bosentan similar to previous paediatric and adult PAH studies of shorter duration.30 In adult PAH patients with Eisenmenger syndrome, bosentan improved exercise capacity and haemodynamics with few AEs, in the placebo-controlled BREATHE-5 trial29 (figure 2, tables 2 and 3). Based on data of the BREATHE-326 (add-on bosentan to prostacyclin therapy in children) and FUTURE-128 studies, the paediatric formulation of bosentan was approved by the EMA for use in children with PAH older than 2 years of age, with a maximum target dose of 2 mg/kg body weight per dose daily (table 2). Recent safety data on the use of bosentan under 2 years of age are available through the FUTURE-3 study on the dosing interval in 64 PAH children from 3 months to 11 years of age. Subsequently, the EMA approved bosentan for the treatment of PAH in children starting from 1 year of age. Elevation of serum liver aminotransferases may occur as a serious AE during bosentan therapy, but was more frequent in adults and children ≥12 years of age (7.8%) than children under 12 years (2.7%) in an European Postmarketing Surveillance study.31 Nevertheless, it is recommended to perform liver function testing monthly in children receiving bosentan.
Ambrisentan
The ARIES-1 and ARIES-2 studies on ambrisentan, an oral selective ETA-receptor antagonist, in adult PAH showed improvement of 6 min walk distance (6MWD) during the 12-week study periods (table 3). Subsequently, a retrospective cohort study on paediatric patients with PAH receiving ambrisentan as add-on therapy or transition from bosentan was conducted. In 38 children with PAH, ambrisentan transition or add-on therapy was associated with an improvement of WHO functional class in 31%. AEs such as headache were frequent, but no elevation of liver enzymes was observed.32
Macitentan
Macitentan, a novel dual ERA, was developed by modifying the structure of bosentan to increase efficacy and safety. Macitentan has improved receptor binding capacity and has fewer drug–drug interactions compared with bosentan. The SERAPHIN study showed that macitentan significantly reduced a composite end point of morbidity and mortality in patients with PAH and also increased exercise capacity.33 No liver toxicity was observed; however, a reduction in blood haemoglobin ≤8 g/dL was found in 4.3% of patients receiving 10 mg macitentan (table 3). In contrast to bosentan, macitentan does not appear to lower the plasma levels of sildenafil, although according specific studies are lacking. Today, no data on the use of macitentan in children are available.
Phosphodiesterase inhibitors (PDE-5i)
Sildenafil
Sildenafil is an orally active inhibitor of phosphodiesterase 5 (PDE-5, among other PDEs), inducing vasodilation and exhibiting antiproliferative effects through the NO/cGMP pathway within the pulmonary vasculature. RCTs in adult patients treated with sildenafil have confirmed favourable results on exercise capacity, symptoms and haemodynamics (table 4). STARTS-139 and STARTS-240 are the first paediatric randomised, placebo-controlled RCTs conducted in treatment-naive children with PAH. In the STARTS-1 trial,39 children with PAH 1–17 years old (≥8 kg body weight) received low-dose (10 mg), medium-dose (10–40 mg) or high-dose (20–80 mg) sildenafil or placebo orally three times daily for the duration of 16 weeks. There were no statistically significant benefits for each sildenafil dosing group versus placebo in terms of the primary outcome measure, peak oxygen consumption (VO2 max.), as assessed by cardiopulmonary exercise testing. However in the subgroup analysis, functional capacity significantly improved in the high-dose sildenafil-group, and PVR index was lowered with medium-dose and high-dose sildenafil. Unfortunately, there was a rise in mortality that was significantly higher in the high-dose sildenafil versus placebo group. These results led to different recommendations by the EMA and the FDA. In 2011, sildenafil received EMA approval for the use in children older than 1 year of age (10 mg three times daily for weight <20 kg, and 20 mg three times daily for weight ≥20 kg). The higher mortality in the high-dose sildenafil group resulted in a warning of the EMA not to use higher doses in 2013. The FDA even released a warning against the (chronic) use of sildenafil in PAH children 1–17 years of age in 2013, which was clarified in 2014 (‘no contraindication’ for paediatric use of sildenafil). Intravenous sildenafil may play a role in children with persistent pulmonary hypertension of the newborn41 and postoperatively after congenital heart surgery;42 ,43 however, midsize, sufficiently powered, prospective studies have not been conducted yet.
Tadalafil
Tadalafil, an oral, selective PDE-5 inhibitor with a longer duration of action than sildenafil, was FDA-approved for use in adults with PAH in 2009. RCTs in adults showed an improvement in exercise capacity and quality of life with the administration of tadalafil (table 4). The use of tadalafil in children with PH has recently increased based on the results of a retrospective study (that suggested clinical efficacy and safety in children with PAH44 and the above-mentioned FDA warning regarding the use of sildenafil). Still, there is no published RCT data on the use of tadalafil in paediatric PAH.
Guanylate cyclase stimulators
Riociguat
Riociguat, an oral agent with dual mode of action that acts in synergy with endogenous NO and also directly stimulates soluble guanylyl cyclase independent of NO availability is another promising therapy. In phase 3 trials on patients with symptomatic PAH (PATENT-1), riociguat therapy led to improved haemodynamics, functional class and time to clinical worsening.45 Subsequently, a subgroup analysis of patients with persistent/recurrent PAH after repair of CHD (n=35) has been performed on the data sets of the PATENT studies.46 Riociguat was well tolerated also in these PAH-CHD patients and improvement of clinical outcomes including 6MWD, PVR, WHO FC and NT-pro B-type natriuretic peptide were consistent with the drug effects in the overall population of PATENT-1 (table 4). An ongoing small trial will describe the use of riociguat in paediatric PAH and particularly its safety and potential advert effects on bone/growth.
Vericiguat
A safety and efficacy phase 2 study on vericiguat (BAY1021189) in adult patients with heart failure and preserved ejection fraction is currently underway (http://www.clinicaltrial.gov; NCT01951638).
Prostacyclin analogues (PCA; prostaglandin I receptor agonists; IP receptor agonists)
Activation of the prostacyclin (PGI2) receptor, IP receptor and prostanoid EP receptors by PGI2 analogues induces vasodilation in the pulmonary circulation, inhibits proliferation of vascular smooth muscle cells (anti-remodelling) and may have anti-inflammatory effects. Inhaled prostacyclin analogues (iloprost, treprostinil) is used in children with PAH, albeit as an off-label therapy, mostly as a sequential combination therapy with an ERA and/or PDE-5 inhibitor. In children and adolescents, particularly the required frequency of inhalations has a negative impact on patient compliance (6–8 times per 24 h for iloprost, 4–6 times per 24 h for trepostinil). In children with progressive, severe PAH (WHO functional class III or IV) and high predictive risk, inadequate response to combination therapy and/or clinical deterioration, (add-on) initiation of subcutaneous treprostinil, intravenous epoprostenol or intravenous treprostinil should be strongly considered (tables 1, 2, 5 and 6 and figures 2, 3).
Epoprostenol (intravenous infusion)
Epoprostenol was the first PGI2 analogue and approved by the FDA for treatment of adults with PAH in 1995. Intravenous epoprostenol improves quality of life and survival in children and adults (non-composite primary outcome) with IPAH23 ,55 ,56 (table 5). In a retrospective analysis, children with IPAH who were treated with intravenous epoprostenol (n=24; AVT non-responders or those with CCB treatment failure) had a 4-year survival rate of 94%23 and a 10-year freedom from death, transplantation or atrial septostomy (AS) of 37% (13/35).22 Combination of intravenous epoprostenol with oral sildenafil, bosentan or both was reported to result in better survival in an UK observational cohort of children with PAH.57 ,58 In children, the effective dose of epoprostenol appears to be higher than in adults. Uptitration of epoprostenol over time is common, but excessive doses (>120 ng/kg/min) may lead to high output states that require downtitration (table 2).59 A new, more chemically stable epoprostenol preparation allows up to once-weekly preparation (up to eight cassettes at one time), which may not require ice packs or special mixing liquids (diluents). However, the short half-life of epoprostenol (2–5 min) keeps patients with PAH at very high risk for pulmonary vascular crisis when there is a sudden problem with drug delivery.
Trepostinil (subcutaneous infusion, intravenous infusion, intermittent inhalation)
Trepostinil is the tricyclic benzidine analogue of epoprostenal that is sufficiently stable to be given continuously per intravenous or subcutaneous route at room temperature in patients with PAH. The FDA approved trepostinil for subcutaneous use in 2002, for intravenous application in 2004 and for inhalative administration in 2009 (for adult PAH RCT results, see table 5). Subcutaneous use of continuous trepostinil via mini pumps is often associated with side pain but allows patients to live free of central venous catheters that inhere the possible complications of line infection, sudden occlusion or extravasal dislocation. Add-on subcutaneous trepostinil has been studied in eight children with PAH at an average dose of 40 ng/kg/min and tolerable side effects.60 Highly efficient intravenous trepostinil can be given by continuous infusion through small, subcutaneous or external pumps, is mixed easily and has a much longer half-life (approximately 4 h) than epoprostenol (table 2). In adults with PAH, the dose of intravenous trepostinil appears to be 2–3 times higher than the dose of epoprostenol.61 A recent retrospective analysis of inhaled trepostinil administration in 29 children with PAH (3–9 breaths, 6 μg/breath, four times daily) showed promising results with an improvement in WHO functional class and 6MWD.62
Iloprost (intermittent inhalation)
The PGI2 analogue iloprost was approved for inhalative treatment of adults with PAH in the USA in 2004. Iloprost can be delivered by simple nebulisation in small children and by adaptive aerosol delivery in older children, adolescents and adults. Because of its short half-life, iloprost should be administered 6–9 times in 12–18 h (every 2–3 h, daily), requiring patient cooperation and solid patient compliance (tables 2 and 5).63 In mid-2015, a new chip and higher concentrated ampoules were released in Europe, so that the inhalation times for iloprost can now be reduced from 10–15 to 5–8 min. The major advantage of inhaled prostacyclin are pulmonary vasodilation, and probably anti-inflammation and anti-remodelling effects, with usually little influence on systemic blood pressure. Headaches, jaw pain and airway reactivity64 may occur at the beginning of iloprost therapy, so that preceding pulmonary function tests are recommended. Inhaled iloprost has been studied in combination with sildenafil65 and bosentan66 in adults, but phase 2/3 studies in children are lacking (table 5).
Beraprost (oral)
Beraprost is a prostacyclin analogue with a short half-life (40 min) that is not available in Europe and the USA, but approved in its long-acting form for PAH therapy in Japan and South Korea.67 While short-term effects of beraprost have been described in adult PAH trials, the efficacy seemed to fade with longer treatment,68 ,69 and paediatric data on the efficacy of beraprost are limited to case reports.
Selexipag (oral)
Selexipag is an oral selective prostacyclin receptor (IP receptor) agonist. Its high functional selectivity for the IP receptor may help minimise gastric side effects. In a pilot RCT in patients with PAH receiving stable ERA and/or PDE-5 inhibitor therapy, selexipag reduced PVR after 17 weeks compared with placebo.70 In the large PAH, event-driven, randomised, double-blind, placebo-controlled phase 3 trial on the efficacy of selexipag on the first morbidity and mortality event (GRIPHON; n=1156),71 selexipag significantly reduced the risk of morbidity/mortality events (composite primary end point: death from any cause or a complication related to PAH up to the end of the treatment period) versus placebo (log-rank p<0.0001) by 40% (HR 0.60; 99% CI 0.46 to 0.78; p<0.001). A primary end point event occurred in 397 patients − 41.6% of those in the placebo group and 27.0% of those in the selexipag group. Disease progression and hospitalisation accounted for 81.9% of the events. There was no significant difference in mortality between the two study groups.71 Oral selexipag may enable earlier combination drug therapy targeting the three-molecular pathways of PAH by combining oral dosing with higher IP receptor selectivity. A drug trial on the use of selexipag in children with PAH is expected to start soon.
Medication used in the intensive care unit (milrinone, levosimendan, inhaled nitric oxide)
Milrinone and levosimendan are lusitropic, positive inotropic agents typically considered for use in patients with PAH in WHO functional class IV. Milrinone, levosimendan and the potent local vasodilator, inhaled nitric oxide, are discussed in a separate article on the treatment of acute PAH in the intensive care unit.3
Combination pharmacotherapy in paediatric PAH
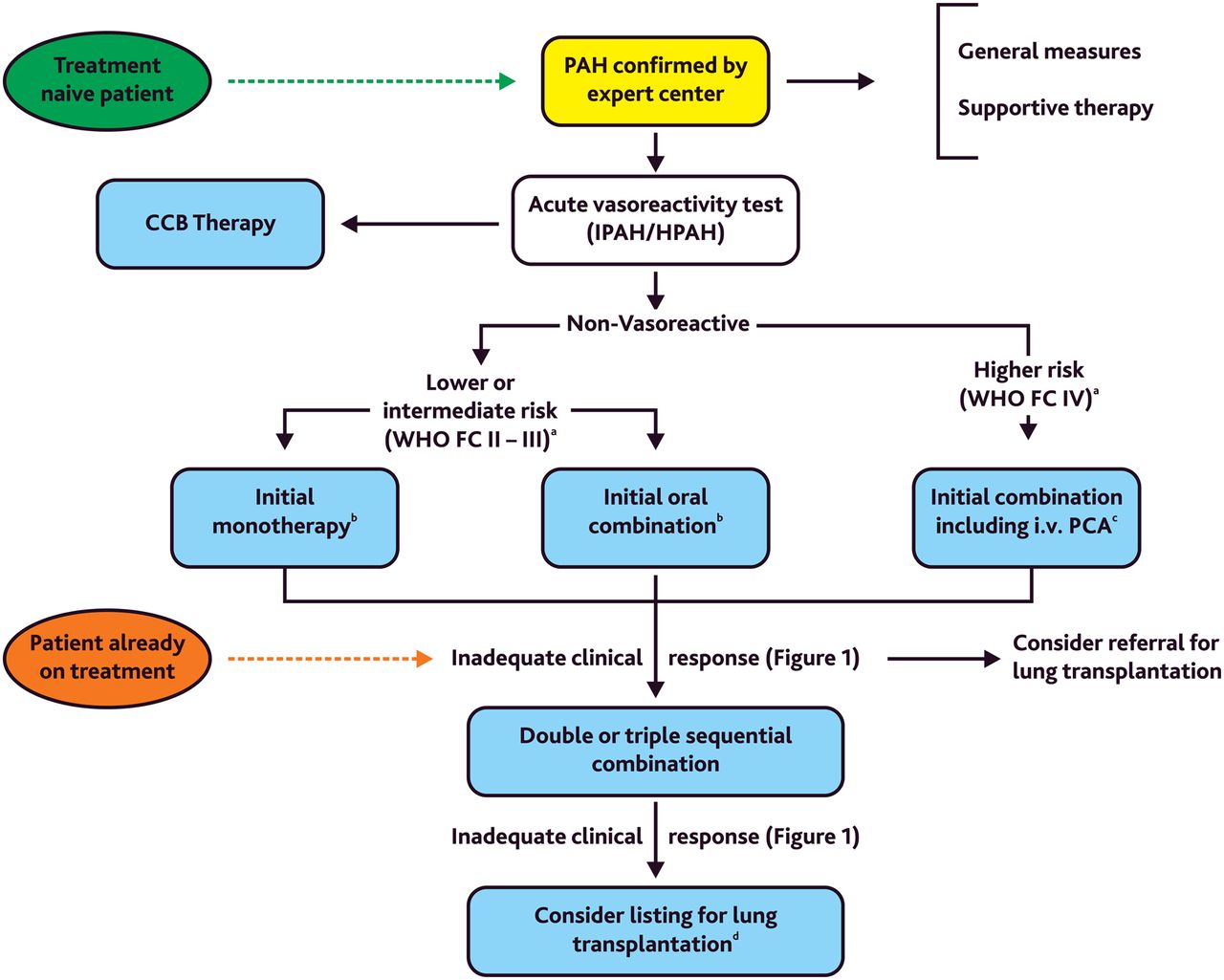
Children who deteriorate on either ERA or PDE-5 inhibitor agents may benefit from early dual or triple combination therapy. Combination therapy (early combination or rapid sequence/add-on) versus monotherapy has not been studied in children yet. Prospective studies are needed to help establish any solid recommendations for combination therapy in children with PH. A modified algorithm on combination therapy based on the adult 2015 ESC/ERS guidelines for PH12 is given in figure 3.

{kind=link}
{kind=link}
{kind=link}
Treatment algorithm for combination therapy in pulmonary arterial hypertension (PAH) (evidenced-based only for adults with PAH, ie, group 1 PH). The ‘intermediate’ risk group is broad and not specifically defined (see figure 1). aSome WHO-FC III patients may be considered high risk (see figure 1, table 1). bIn adult PAH, initial combination with ambrisentan plus tadalafil has been shown to be superior to initial monotherapy with ambrisentan or tadalafil in delaying clinical failure. cIntravenous epoprostenol should be prioritised as it has reduced the 3 months rate for mortality in high-risk adult patients with PAH also as monotherapy. dConsider also balloon atrial septostomy or Potts shunt. Modified from Galiè et al.12 CCB, calcium channel blockers; DPAH, drug-induced PAH; HPAH, heritable PAH; IPAH, idiopathic PAH; IV., intravenous; PCA, prostacyclin analogues; WHO-FC, WHO functional class.
Characteristics and major findings of RCTs testing upfront combination therapy in adult PAH are shown in table 6.
Drug interactions
The potentially significant drug interactions with PAH-targeted therapies are listed in table 7.
Potentially significant drug interactions with PAH targeted therapies
Experimental compounds and strategies
Because of the lack of any validated prospective paediatric data or even drug approval in childhood, the above-mentioned compounds macitentan, tadalafil, riociguat, vericiguat, beraprost and selexipag must be considered experimental paediatric PH pharmacotherapy (‘compassionate use’) until convincing clinical data become available. Venoarterial-ECMO as bridge to lung transplantation or bridge to recovery in severe PAH is discussed in a separate article of the European Paediatric Pulmonary Vascular Disease Network.3 A limited number of studies on the value of balloon atrial septostomy (AS) and Potts shunt in advanced paediatric PAH are available and will be discussed in the following section.
Catheter-based and surgical therapy of advanced PAH
To avoid overt right heart failure in patients with severe and refractory PAH, a right-to-left shunt can be created with the aim to decompress right heart structures and to increase and maintain cardiac output. With these interventions, a haemodynamic situation occurs, which is similar to the physiology of Eisenmenger patients, whose RV systolic function is preserved longer, and who have somewhat better survival than IPAH patients without a decompressing shunt.88 Treatment of PAH associated with CHD is discussed in a separate article of the European Paediatric PVD Network.4
Atrial septostomy
Atrial septostomy (AS) is the most frequently applied interventional technique that is used as a palliative therapy in the management of patients with advanced PH and failing RV89 when medical therapy has failed. AS improves symptoms and quality of life in paediatric PAH and may serve as bridge to lung transplantation. Following puncture of the interatrial septum in Brockenbrough technique, the defect is stepwise balloon-dilated until the predetermined diameter is achieved or the arterial oxygen saturation has dropped 10% from baseline. As spontaneous closure of the created defect occurs in almost 10%, the procedure may be repeated and/or a stent device can be implanted to keep the communication patent.
According to a 2009 worldwide retrospective analysis of 223 patients including a significant number of children, procedure-related mortality was very high (7.1% at 24 h and 14.8% at 1 month),90 while immediate and long-term haemodynamic response including survival (available in 128 patients) was encouraging with a median survival of 60 months.90
However, more recent studies suggest that AS can be a safe procedure in selected patients with PAH when performed in expert centres, and that AS in combination with PAH-targeted therapy improves survival: a recent retrospective study from Mexico analysed the clinical impact of AS in treatment-naive patients with severe PAH (age range 22–62 years, 85% IPAH: 21 underwent AS alone, 11 received additional drug therapy).91 There was one procedure-related death in 50 procedures performed on 32 patients. Median survival was 53 months in the AS-alone group and 83 months in the AS+PAH-targeted therapy group.91 In a recent retrospective analysis of 46 AS procedures performed in 32 paediatric/adult patients with PAH (69% in WHO FC III or IV) in a high-volume PH centre in New York between 2002 and 2013, there were no procedural complications or deaths. Seven of 32 patients were successfully bridged to lung transplantation. Lung transplantation-free and repeat-AS-free survival at 30 days, 1 year and 5 years was 87%, 61% and 32%, respectively.92
Based on the risk factors found in the international study with high procedure-related mortality,90 contraindications for AS include (1) a mean right atrial pressure of >20 mm Hg, (2) resting arterial oxygen saturation <90%, (3) severe RV failure and (4) patients with impending death. AS can be considered in patients in functional class III and IV and recurrent syncope under combined medical therapy, and as palliative bridge to transplant increasing the chance for survival while waiting for a donor organ.92 ,93
Potts shunt (LPA-DAO)
This surgical procedure implies the creation of a connection between the left pulmonary artery (LPA) and the descending aorta (DAO), which allows right-to-left shunting, similarly to a patient with a patent ductus arteriosus-related Eisenmenger syndrome. The use of a Potts (LPA-DAO) shunt in suprasystemic PH is considered advantageous compared with AS as it provides high oxygen saturated blood for the coronary arteries and the central nervous system and only causes desaturation of the lower body. In addition, the risk of fatal paradoxical embolisms may be lower compared with a connection on the atrial level. Another benefit arises from its effect on haemodynamics by the relief of RV pressure overload in systole and, in part, also in diastole with a subsequent reduction in shifting of the interventricular septum towards the LV with an improvement in systolic and diastolic LV performance. The connection between the pulmonary artery and the descending aorta can be achieved either by a direct side-by-side anastomosis or by using a synthetic graft tube/prosthesis, which should have the size of the descending aorta to allow sufficient decompression of the RV. A run-off through the Potts shunt with decreased pulmonary perfusion and extreme desaturation of the lower body with subsequent undersupply of the myocardium and the brain should be avoided.
Given that the experience with the Potts shunt procedure is nearly exclusively available in children, these data cannot be extrapolated to severely ill adults, which may have a considerably higher periprocedural risk. The procedure may be considered in patients with suprasystemic PH refractory to any medical treatment including combined therapy presenting in New York Heart Association/WHO functional class IV.
The largest series published by Baruteau and colleagues consisted of 24 children with drug-refractory PAH in which a permanent Potts shunt was created (19 surgical LPA-DAO, 6 via stenting of a persistent ductus arteriosus (PDA)).94 Six patients experienced severe postoperative complications and three early deaths relating to low cardiac output occurred. After a median follow-up of 2.1 years, the 21 survivors showed persistent improvement in functional capacities, and none of the patients had syncope or overt RV failure.94 These favourable long-term results suggest that creation of a Potts shunt can be a valuable alternative, or bridge to bilateral lung transplantation, at least in selected cases.
Recently, several case series demonstrated the feasibility of the pure catheter-based interventional implementation of the connection between the left PA and the descending aorta.95–98 The obviously most elegant method is the implantation of a stent in a still patent PDA, which is not infrequently present in infants and young children.95–97 This procedure is an established method in CHD with duct-dependent circulation and can be established with considerable low periprocedural risk in experienced centres. Currently, the interventional de novo creation of a LPA-DAO connection with a covered stent from the LPA96 or DAO side98 has been shown to be feasible but currently must be considered a high-risk procedure in patients with end-stage PAH who are too sick to undergo surgery.
Pulmonary artery denervation
The rationale for the pulmonary artery denervation (PADN) procedure is based on the observation that baroreceptors and sympathetic nerve fibres are localised in or near the bifurcation of the main pulmonary artery. Two small pilot studies demonstrated some benefits of PADN on exercise capacity and symptoms of patients with adult PAH at 3 and 12 months follow-up.99 ,100 Data on the safety and efficacy of PADN in children with PAH are lacking.
Challenges in clinical studies of paediatric PAH
Identification of valid treatment goals in paediatric PAH.101 ,102
Regulatory requirements, patient recruitment and retention, clinical trial end points for paediatric PAH trials.102
Need for interdisciplinary, international PVD networks to conduct multicentre studies and to establish registries.103 ,104
Initiation of a prospective multicenter study on early combination therapy in paediatric PH including a comparative group (early combined dual or triple combination, rapid sequence of two agents).
When and how to perform a Potts shunt procedure for advanced PAH (surgery, intervention?) and how to combine this pressure-unloading shunt with combination pharmacotherapy?
Initiation of investigator-initiated pilot and/or industry-sponsored phase 2–4 studies on the safety and efficacy of new compounds recently published/approved for adult PAH (macitentan, riociguat, selexipag, trepostinil).
Acknowledgments
The authors thank Damien Bonnet, Paris, for critically reviewing this manuscript and this manuscript, and Cengiz Tamak, Hannover, for editorial assistance.
References
Footnotes
This paper is a product of the writing group of the European Paediatric Pulmonary Vascular Disease (PVD) Network (Writing Group Chair: G. Hansmann, Writing Group Co-Chair: C. Apitz). ISHLT, International Society of Heart and Lung Transplantation; DGPK, German Society of Paediatric Cardiology.
Funding GH currently receives grant support from the German Research Foundation (DFG; HA 4348/2-1), Kinderherzen e.V. (W-H-001-2014), and Stiftung Kinderherz (2511-6-13-011). CA currently receives grant funding from Stiftung Kinderherz (2511-10-13-001) and Behring-Röntgen-Stiftung (59-0018).
This Heart supplement was produced with support from an unrestricted educational grant from Actelion Pharmaceuticals Germany GmbH, Bayer Pharma AG, and Pfizer Inc. None of these organisations had any influence on the composition of the writing group or the content of the articles published in this supplement. Open Access publication of this article was sponsored by Actelion Pharmaceuticals Germany GmbH.
Competing interests GH and CA declare no conflict of interest.
Provenance and peer review Not commissioned; externally peer reviewed.