Article Text

Abstract
Objective: To investigate the effects of short term atorvastatin treatment on forearm vasodilatory response to reactive hyperaemia (RH%) and on components of the thrombosis–fibrinolysis system (antithrombin III, proteins and S, factors V and VII, von Willebrand factor, tissue plasminogen activator (tPA), and plasminogen activator inhibitor (PAI-1)) in patients with heart failure.
Patients and methods: 35 patients with heart failure were enrolled in this study; 17 patients received atorvastatin 10 mg/day and 18 patients received no statin for four weeks. Forearm blood flow (FBF) was measured by venous occlusion strain gauge plethysmography. RH% and forearm vasodilatory response to nitrate were defined as the percentage change of FBF from rest to the maximum flow during reactive hyperaemia and after nitrate administration, respectively. Plasma concentrations of antithrombin III, protein C, protein S, factor V, factor VII, von Willebrand factor, tPA, and PAI-1 were determined before and after treatment.
Results: Maximum hyperaemic FBF remained unchanged in both groups. Baseline FBF was slightly but not significantly decreased in the atorvastatin treated group. RH% was significantly increased only in the atorvastatin treated group, from mean (SD) 42.44 (18.9)% to 83.7 (36.1)% (p < 0.01). Plasma concentrations of antithrombin III (from mean (SD) 81.7 (11.37)% to 73.5 (13.8)%), protein C (from mean (SD) 88.3 (26.9)% to 63.9 (25.0)%), factor V (from mean (SD) 126.2 (33.4)% to 94.9 (29.8)%), tPA (from median (25th–75th percentile) 11.68 (8.60–20.95) ng/ml to 10.30 (8.65–15.12) ng/ml), and PAI-1 (from median (25th–75th percentile) 3.10 (2.15–4.40) IU/l to 1.90 (0.75–3.0) IU/l) were significantly decreased in the atorvastatin treated group (p < 0.05) but not in the control group. Plasma concentrations of von Willebrand factor, factor VII, and protein S remained unaffected in both groups.
Conclusion: Atorvastatin did not change the maximum hyperaemic flow, although it decreased plasma concentrations of antithrombin III, protein C, factor V, tPA, and PAI-1 in patients with heart failure. Therefore, short term treatment with atorvastatin may affect the expression of both endothelium and liver derived components of the thrombosis–fibrinolysis system in patients with heart failure.
- eNOS, endothelial nitric oxide synthase
- FBF, forearm blood flow
- ELISA, enzyme linked immunosorbent assay
- NTG%, forearm vasodilatory response to nitrate
- PAI-1, plasminogen activator inhibitor 1
- RH%, forearm vasodilatory response to reactive hyperaemia
- TNFα, tumour necrosis factor α
- tPA, tissue plasminogen activator
- heart failure
- statins
- endothelium
- thrombosis
- fibrinolysis
Statistics from Altmetric.com
- eNOS, endothelial nitric oxide synthase
- FBF, forearm blood flow
- ELISA, enzyme linked immunosorbent assay
- NTG%, forearm vasodilatory response to nitrate
- PAI-1, plasminogen activator inhibitor 1
- RH%, forearm vasodilatory response to reactive hyperaemia
- TNFα, tumour necrosis factor α
- tPA, tissue plasminogen activator
Congestive heart failure is characterised by left ventricular dysfunction and reduced exercise capacity. It is associated with activation of neurohumoral systems, fluid accumulation, limitation of peripheral circulation, and end organ failure.1,2
Evidence suggests that lipids are associated with increased thrombogenicity and mortality in patients with advanced atherosclerosis.3 Statin treatment has been shown to reduce mortality in patients with coronary artery disease,3–6 to restore endothelial function in patients with hypercholesterolaemia and with coronary atherosclerosis,7 and to ameliorate the inflammatory and thrombotic process in patients with advanced atherosclerosis.8 However, the effects of statins on the vascular endothelium and thrombosis–fibrinolysis system in patients with heart failure are unclear. The purpose of this study was to investigate the effects of short term treatment with atorvastatin on endothelial function and on both endothelium and liver derived components of the thrombosis–fibrinolysis system, specifically antithrombin III, proteins C and S, factors V and VII, von Willebrand factor, tissue plasminogen activator (tPA), and plasminogen activator inhibitor 1 (PAI-1), in patients with heart failure.
METHODS
Patients
Thirty five patients with heart failure were enrolled in this study. All patients had clinical evidence of heart failure and were in New York Heart Association functional classes II–IV, with left ventricular ejection fraction < 35% as assessed by contrast ventriculography and fasting cholesterol concentrations < 5.7 mmol/l. All patients had been in a stable clinical state for at least three months before study entry. Exclusion criteria were left ventricular hypertrophy, overt atherosclerotic peripheral vascular disease, acute and chronic inflammatory diseases involving organs other than the heart (for example, liver diseases), other cardiac disease, smoking, use of lipid lowering agents during the previous six months, atrial fibrillation, and use of antithrombotic agents other than aspirin. Table 1 presents the patients’ baseline characteristics.
Baseline characteristics and medication
Protocol
Patients were randomly allocated to two groups by an adaptive dynamic random allocation method and received atorvastatin 10 mg/day (n = 17, mean (SD) age 63 (12.5) years, 12 with ischaemic and five with non-ischaemic heart failure) or no statin (control group, n = 18, mean (SD) age 67 (9.9) years, 13 with ischaemic and five with non-ischaemic heart failure) for four weeks. All patients were treated with diuretics and angiotensin converting enzyme inhibitors. Table 1 presents the participants’ medication. Forearm blood flow (FBF) was measured by gauge strain plethysmography (EC-400, D E Hokanson, Inc, Bellevue, Washington, USA) as previously described.9,10 Before measurements were started, patients rested in a supine position for 30 minutes. FBF output signal was transmitted to a personal computer (Hokanson NIVP3 software). FBF was finally calculated as the percentage change of arm volume/100 ml tissue/min. Reactive hyperaemia was produced by the release of the ischaemia cuff after four minute ischaemia of the distal forearm. Forearm vasodilatory response to reactive hyperaemia (RH%) was defined as the percentage change of FBF from baseline to the maximum flow during reactive hyperaemia relative to baseline. Forearm vasodilatory response to nitrate (NTG%) was expressed as the percentage change of flow from baseline to the maximum flow after sublingual administration of 0.4 mg glyceryl trinitrate. RH% was regarded as an index of endothelial function in forearm resistance vessels and NTG% as an index of endothelium independent dilatation, as previously described.9–11
Venous blood samples were collected at the beginning of the study and after four weeks of treatment. At the second visit, patients were asked to take the last dose of intervention compound 12 hours before the beginning of the protocol. The protocol was approved by the research ethics committee and each patient gave written informed consent. All measurements were taken in the morning in a dark, quiet room under constant temperature (22–25°C).
Blood sample measurements
Venous blood samples were taken before plethysmography was performed. After centrifugation at 3500 rpm at 4°C for 15 minutes, plasma or serum was collected and stored at −80°C until assayed. Routine chemical methods were used to determine serum concentrations of total cholesterol, high density lipoprotein, triglycerides, and glucose. Enzyme linked immunosorbent assay (ELISA) was used to determine plasma concentrations of von Willebrand factor (Dade Behring Inc, Liederbach, Germany) and tPA antigen (Asserchrom, Diagnostica Stago, Asnières, France). Clotting assays were used to determine factor V (Dade Behring), factor VII (Dade Behring), and protein S (Stalcot protein S kit, Diagnostica Stago). Standard chromogenic substrate techniques were used to determine protein C (Dade Behring), PAI-1 activity (Dade Behring), and antithrombin III activity (Berichrom, Dade Behring).
Statistical analysis
Statistical analysis was performed with a personal computer and the SPSS 11.0 statistical package (SPSS Inc, Chicago, Illinois, USA). All continuous variables were tested for normal distribution by the Kolmogorov-Smirnov test. Normally distributed variables were expressed as mean (SD) and non-normally distributed variables were expressed as median (25th–75th percentile). Unpaired t test with two tails was used to evaluate the differences in normally distributed variables between the two groups at baseline, and Mann-Whitney U test was used to compare non-normally distributed variables. Paired samples t test or Wilcoxon ranks test were used to evaluate the change of each normally or non-normally distributed parameter, respectively, after treatment. Changes in the two groups were compared by two way analysis of variance for repeated measurements. A probability value of p < 0.05 was regarded as significant.
RESULTS
All the examined parameters remained unchanged in the control group (table 2). There were no significant differences in baseline values of all the examined parameters between the two groups. Serum cholesterol and triglyceride concentrations were significantly decreased in the atorvastatin treated group (p < 0.01 for both) but were unaffected in the control group (table 2).
Effects of treatment with atorvastatin or no statin (control group) on forearm blood flow, components of the thrombosis–fibrinolysis system, and serum lipids in patients with heart failure at baseline and after four weeks
Effects of atorvastatin on endothelial function
FBF at rest was unchanged in the control group but decreased slightly but not significantly in the atorvastatin treated group after treatment (table 2). Similarly, maximum hyperaemic FBF was unchanged in both the control and atorvastatin treated groups (table 2). However, RH% was significantly increased in the atorvastatin treated group (p < 0.01) but was unaffected in the control group (not significant) (table 2, fig 1). The change of RH% was significantly greater in the atorvastatin treated group (41.3%, 95% confidence interval (CI) 21.6 to 60.9) than in the control group (−3.24%, 95% CI −12.7 to 6.3; p < 0.001). Atorvastatin treatment had no effect on NTG% (table 2).

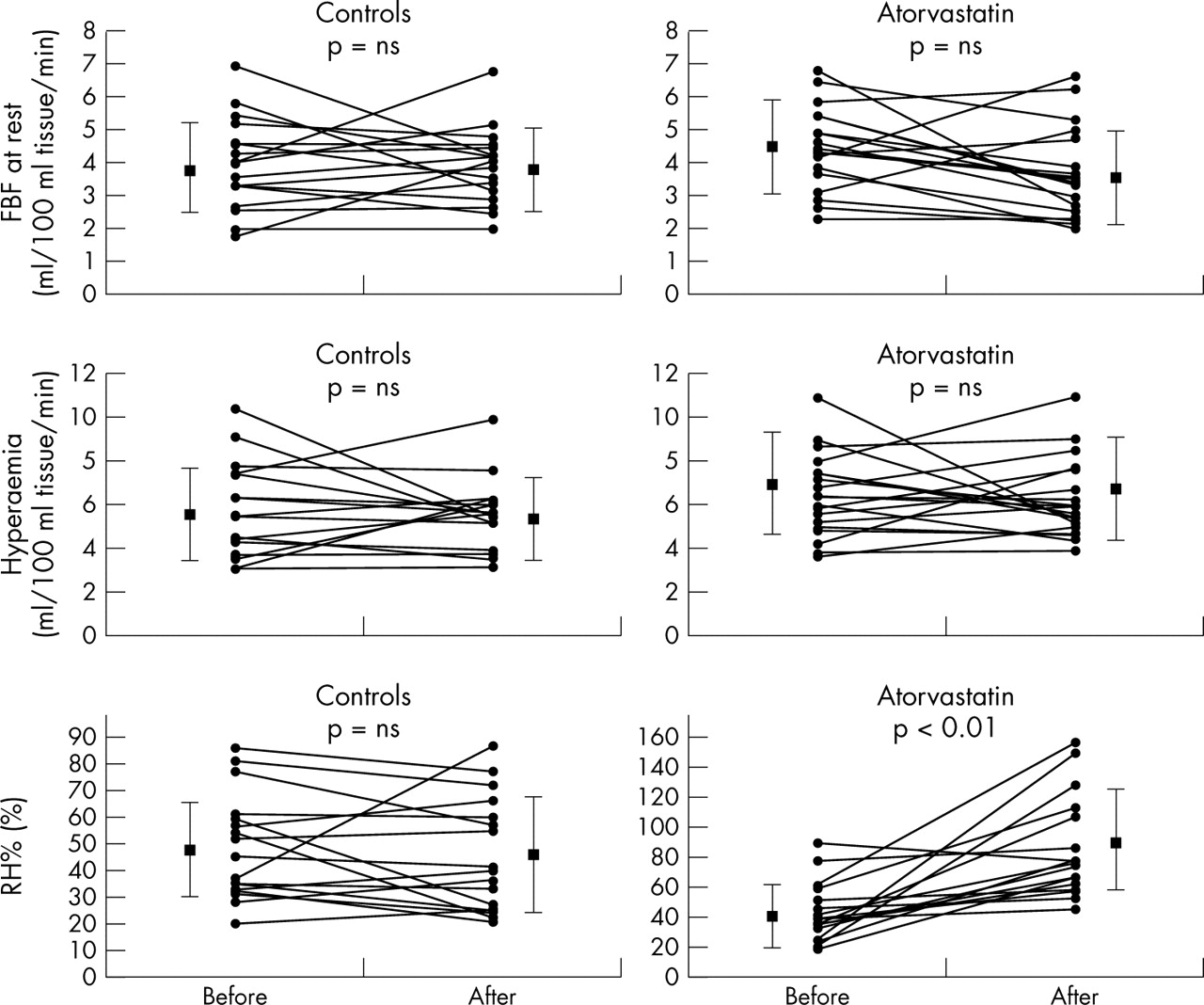{kind=link}
Effect of atorvastatin treatment on forearm blood flow (FBF), maximum hyperaemic flow (hyperaemia), and forearm vasodilatory response to reactive hyperaemia (RH%) (individual data). FBF and hyperaemic flow remained unchanged in both atorvastatin treated and control groups. RH% increased significantly in the atorvastatin treated group (p < 0.01) but remained unchanged in the control group. Data are mean (SD). NS, not significant.
Effects of atorvastatin on the thrombosis–fibrinolysis system
Plasma concentrations of PAI-1 were decreased in the atorvastatin treated group (p < 0.05) and were slightly increased in the control group (not significant) (table 2). The change of PAI-1 was significantly greater in the atorvastatin treated group (−1.1 IU/l, 95% CI −0.7 to −2.0 IU/l) than in the control group (0.6 IU/l, 95% CI −0.1 to 1.3 IU/l; p < 0.01). Plasma antithrombin III concentrations were decreased in the atorvastatin treated group (p < 0.01) but were slightly increased in the control group (not significant) (table 2). The change in antithrombin III in the atorvastatin treated group (−8.2%, 95% CI −13.8% to 2.6%) was significantly different from the change in the control group (2.7%, 95% CI −6.3% to 11.7%; p < 0.05). Protein C concentrations were significantly decreased in the atorvastatin treated group (p < 0.01) but were unchanged in the control group (not significant) (table 2). The change in protein C concentrations was significantly higher in the atorvastatin treated group (−24.4%, 95% CI −13.2% to −35.6%) than in the control group (−0.3%, 95% CI −8.9% to 8.3%; p < 0.01). Concentrations of factor V were also significantly decreased in the atorvastatin treated group only (p < 0.01) (table 2); the change in the atorvastatin treated group (−31.2%, 95% CI −16.8% to −45.7%) was also significantly greater than the change in the control group (−5.6%, 95% CI −23.6% to 12.4%; p < 0.05). Similarly, plasma concentrations of tPA were significantly decreased in the atorvastatin treated group (p < 0.05) but were unchanged in the control group (not significant). Plasma concentrations of factor VII, von Willebrand factor, and protein S were unchanged in both the atorvastatin treated and the control groups (table 2).
DISCUSSION
The results of this study indicate that short term treatment with atorvastatin did not affect the maximum hyperaemic flow in the forearm, although it increased the percentage change of flow during reactive hyperaemia. Furthermore, atorvastatin decreased plasma concentrations of tPA, PAI-1, antithrombin III, protein C, and factor V in patients with heart failure. These findings support the theory that statin treatment may affect the thrombosis–fibrinolysis system in these patients.
Endothelial function and thrombotic mechanisms in heart failure
Heart failure is associated with endothelial dysfunction as a result of increased oxidative stress status, reduced blood flow, and decreased shear stress stimuli.2 Other mechanisms such as increased concentrations of cytokines (such as tumour necrosis factor α (TNFα))12 and increased angiotensin converting enzyme activity may also lead to endothelial dysfunction in heart failure.12,13
Heart failure is accompanied by a so called hypercoagulative state as a result of impaired platelet function, impaired endothelial function, and coagulation indices.14 Increased concentrations of von Willebrand factor,15,16 a marker of endothelial cell integrity, and thrombin-antithrombin III complex concentrations17 are observed in patients with heart failure. Furthermore, increased concentrations of endothelium derived components of the fibrinolysis system, such as tPA2 and PAI-1, have also been documented in these patients.2,14,18 Proinflammatory cytokines such as TNFα15 may be involved in the disturbance of the procoagulant–fibrinolytic balance stimulating both the coagulation16,19 and fibrinolysis systems, leading to possible microvascular thrombosis.14,19
Effects of statins on the vascular endothelium and thrombosis–fibrinolysis system
The first evidence for an association between plasma lipids and coagulation factors was provided by the Northwick Park heart study,20 which reported a positive correlation between serum lipids and factor VII. Furthermore, serum lipids (especially triglyceride-rich lipoprotein) are associated with fibrinogen, PAI-1, and activated factor VII concentrations.21
Statins and endothelium derived components of the thrombosis–fibrinolysis system
Statins may affect thrombus formation and concentrations of fibrinogen, PAI-1, and other thrombotic molecules in patients with hypercholesterolaemia.7 Simvastatin reduces the expression of PAI-1 and stimulates the expression of tPA in human vascular smooth muscle and endothelial cells.22 The net result of these two effects is to greatly tilt the fibrinolytic balance in the vessel wall and to increase fibrinolytic activity.7,23 It has also been shown that lipid lowering treatment significantly decreases thrombus formation on an injured vascular surface under dynamic flow conditions and improves the fibrinolytic profile in patients with hypercholesterolaemia with or without coronary artery disease24 and reduces thrombin generation in men with hypercholesterolaemia.25 These antithrombotic effects of statins can synergise with the antiplatelet aggregating effect of nitric oxide, which occurs in response to statins.26,27 Evidence suggests that statins increase basal endothelium nitric oxide release27 by activating transcription of the endothelial nitric oxide synthase (eNOS) gene in human endothelial cells in vivo28; this may have antithrombotic effects. Beyond their effect on the expression of eNOS,29,30 statins may also stabilise eNOS mRNA inside endothelial cells and prevent oxidative deactivation of nitric oxide.31 It has been shown that statins also have antioxidant and anti-inflammatory properties,31,32 thus improving endothelial function.32
In the present study, FBF at rest, maximum hyperaemic flow, and NTG% were unchanged in both the atorvastatin treated and the control groups, indicating that statin treatment does not directly affect forearm resting tone or the underlying vascular smooth muscle cells. Furthermore, atorvastatin decreased plasma concentrations of tPA and PAI-1, implying that statins have a beneficial effect on endothelial cell integrity. However, we did not find any effect of atorvastatin on plasma concentrations of von Willebrand factor (a finding compatible with previous observations)33 showing that, despite the beneficial effect of atorvastatin on most of the endothelium derived components of the thrombosis–fibrinolysis system, atorvastatin may not affect plasma concentrations of von Willebrand factor.
Atorvastatin and liver derived components of the thrombosis–fibrinolysis system
Atorvastatin seems to have anti-inflammatory properties, by decreasing serum concentrations of proinflammatory cytokines (such as interleukin 1b, interleukin 6, or TNFα) and by modifying the acute phase response.34 Furthermore, it has a direct effect on hepatocyte function and biosynthetic ability.31,32 It has been shown that statin treatment may affect blood clotting formation in patients with advanced atherosclerosis.35,36 However, the effect of statins on hepatocyte derived components of the coagulation–fibrinolysis system has not been evaluated until now. In the present study we found a significant decrease in plasma concentrations of antithrombin III, protein C, and factor V, possibly as a result of the effect of atorvastatin on the biosynthetic ability of hepatocytes. Furthermore, atorvastatin may depress both the fibrinolysis and coagulation systems through the decrease of proinflammatory cytokines such as TNFα,15,19 which stimulate the expression of parameters from both systems.14,15,19 However, it is unknown whether atorvastatin finally promotes thrombosis or fibrinolysis in these patients, since it decreases the production of liver derived components of both systems. Limitations of the present study were the lack of a placebo treated control group and the relatively small number of patients, although our results reached significance.
Conclusions
This is the first study evaluating the effect of atorvastatin on endothelial function and the thrombosis–fibrinolysis system in patients with heart failure. Atorvastatin treatment for four weeks did not affect the maximum hyperaemic flow in the forearm, although it affected the coagulation and fibrinolysis systems by reducing plasma concentrations of antithrombin III, protein C, factor V, tPA, and PAI-1.