Article Text

Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterised by fibrofatty replacement of predominantly the right ventricle and high risk of ventricular arrhythmias and sudden cardiac death (SCD). Early diagnosis and accurate risk assessment are challenging yet essential for SCD prevention. This manuscript summarises the current state of the art on ARVC diagnosis and risk stratification. Improving the 2010 diagnostic criteria is an ongoing discussion. Several studies suggest that early diagnosis may be facilitated by including deformation imaging (‘strain’) for objective assessment of wall motion abnormalities, which was shown to have high sensitivity for preclinical disease. Adding fibrofatty replacement detected by late gadolinium enhancement or T1 mapping in cardiac MRI as criterion for diagnosis is increasingly suggested but requires more supporting evidence from consecutive patient cohorts. In addition to the traditional right-dominant ARVC, standard criteria for arrhythmogenic cardiomyopathy (ACM) and arrhythmogenic left ventricular cardiomyopathy (ALVC) are on the horizon. After diagnosis confirmation, the primary management goal is SCD prevention, for which an implantable cardioverter-defibrillator is the only proven therapy. Prior studies determined that younger age, male sex, previous (non-) sustained ventricular tachycardia, syncope, extent of T-wave inversion, frequent premature ectopic beats and lower biventricular ejection fraction are risk factors for subsequent events. Previous implantable cardioverter-defibrillator indication guidelines were however limited to three expert-opinion flow charts stratifying patients in risk groups. Now, two multivariable risk prediction models (arvcrisk.com) combine the abovementioned risk factors to estimate individual risks. Of note, both the flow charts and prediction models require clinical validation studies to determine which should be recommended.
- arrhythmogenic right ventricular dysplasia
- risk factors
- diagnostic imaging
- defibrillators
- implantable
- genetic diseases
- inborn
Statistics from Altmetric.com
- arrhythmogenic right ventricular dysplasia
- risk factors
- diagnostic imaging
- defibrillators
- implantable
- genetic diseases
- inborn
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a familial disease characterised by fibrofatty replacement of predominantly right ventricular (RV) myocardium, ventricular arrhythmias, sudden cardiac death (SCD) and/or heart failure. While the first historical description was in 1763 by Giovanni Maria Lancisi in De Motu Cordis et Aneurysmatibus, Dr Marcus was the first to describe ARVC in modern literature.1 Now, after four decades of research in electrophysiology, molecular genetics and cardiac imaging, much has changed in our understanding and clinical management of ARVC.
While originally classified as dysplasia (ie, developmental birth defect), we now recognise ARVC as a genetic cardiomyopathy with an autosomal dominant inheritance pattern with incomplete penetrance. The rise of cardiogenetic clinics enabled cascade screening of relatives identifying those at risk of developing ARVC. The at-risk population started growing rapidly, resulting in a noticeable shift in the clinical population from patients with overt disease towards asymptomatic patients with little or no disease expression. This urged clinical management to focus on early disease detection and risk prediction, while guidance from research and guidelines was limited. As a first step, the original 1994 diagnostic ‘Task Force Criteria’ (TFC) were revised in 2010 to improve sensitivity for early and familial disease.2
Despite the revised criteria, many clinical challenges remain, which mainly result from incomplete penetrance and highly variable disease expression. In this review, we provide an overview of the state of the art in pathophysiology, genetics and management of ARVC and focus on the recent developments in diagnosis and risk stratification.
ARVC, arrhythmogenic left ventricular cardiomyopathy (ALVC) and arrhythmogenic cardiomyopathy (ACM): what is in a name?
Over the years, several terms were introduced related to this disease (figure 1). The original term arrhythmogenic right ventricular dysplasia (ARVD) refers to the developmental disorder (‘dysplasia’) that this disease was thought to be at the time.1 With increasing knowledge, ARVD was recognised as a progressive disease that developed after birth (‘cardiomyopathy’) leading to its replacement by the more correct term ARVC3; hence, ARVD, ARVC or ARVD/C can be considered synonyms. These terms relate to our most classic understanding of this disease: predominant RV involvement, fulfilment of the 2010 TFC and pathogenic variants in desmosomal genes.
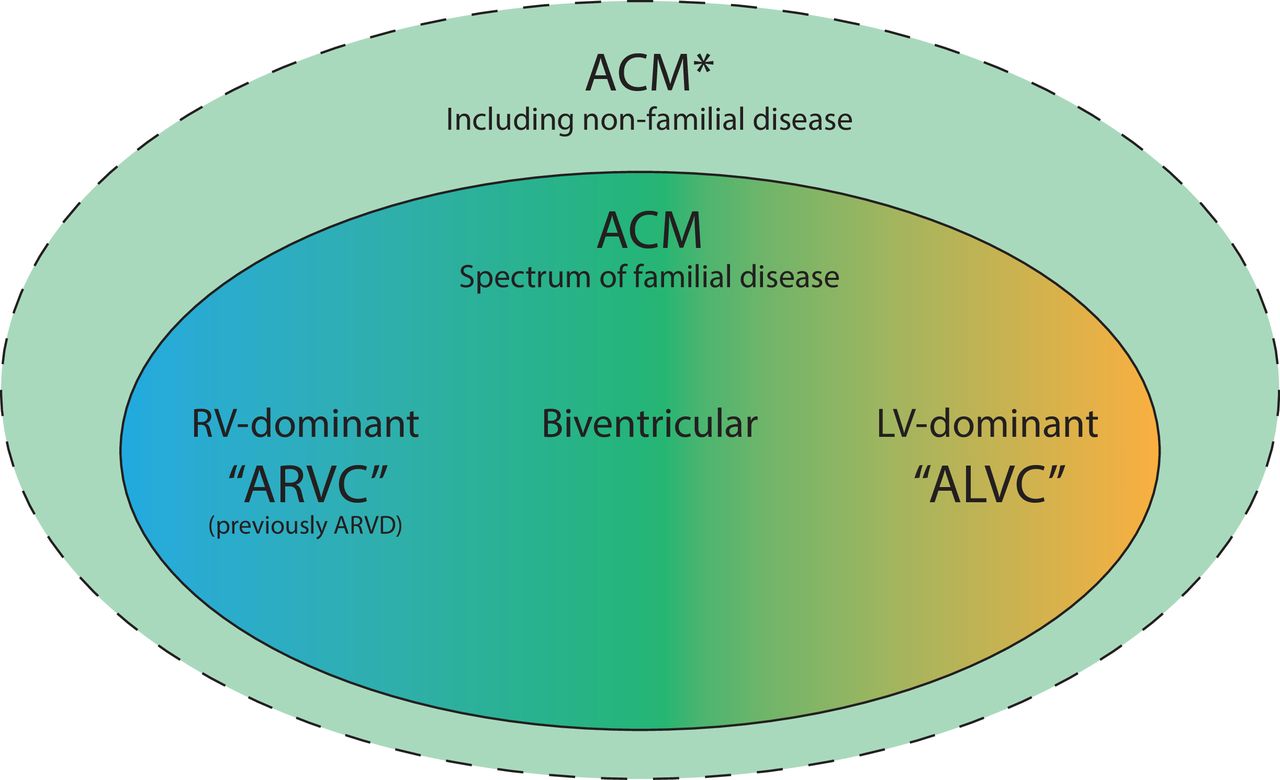
Schematic representation of terminology: ARVC, ALVC and ACM. Arrhythmogenic right ventricular cardiomyopathy (ARVC) refers to the most classical right ventricular (RV) dominant concept of this familial cardiomyopathy characterised by fibrofatty replacement of the myocardium predisposing to ventricular dysfunction and arrhythmias, and arrhythmogenic left ventricular cardiomyopathy (ALVC) in case of left ventricular (LV) dominant disease. Arrhythmogenic cardiomyopathy (ACM) refers of the entire spectrum of ARVC, ALVC and biventricular phenotypes, but some literature includes non-familial diseases in the ACM definition as well. *The inclusion of non-familial disease such as inflammatory (eg, sarcoidosis) or infectious (eg, Chagas disease) is subject of debate.
While almost all ARVC patients show some degree of left ventricular (LV) involvement, a proportion of patients has predominant LV disease.4 Since this does not fit the classical ARVC concept, the terms left-dominant arrhythmogenic cardiomyopathy or ALVC were introduced. ALVC occurs more frequently with DSP and non-desmosomal (eg, PLN and LMNA) gene variants.4 5 However, most gene variants are observed in both ARVC and ALVC patients.
To cover the whole spectrum of biventricular involvement, the term arrhythmogenic cardiomyopathy (ACM or AC) was introduced to describe this familial disease with a common genetic background.6 At present, however, a uniform definition of ACM remains absent: the range of diseases designated as ACM varies from classical ARVC to almost any arrhythmogenic myocardial disorder. To most, it seems obvious to restrict the definition to familial disease, ensuring a similar aetiology.7–9 However, some define ACM as any arrhythmogenic disorder of the myocardium not secondary to ischaemic, hypertensive or valvular disease, thereby including infectious and inflammatory diseases (eg, Chagas and sarcoidosis).10 Nonetheless, it can be appreciated that every ARVC is considered ACM, but not every ACM is ARVC. For clarity, we focus on familial/genetic disease with predominant RV involvement (ie, ‘ARVC’) throughout the remainder of this manuscript.
Epidemiology and clinical presentation
The estimated population prevalence of ARVC ranges from 1:5000 to 1:2000,11 although under-recognition is probably an important problem. Affected patients typically present between the ages of 20–40 years, with symptoms ranging from palpitations, (pre-)syncope, to even SCD as first manifestation.12 On clinical evaluation, ARVC can be categorised into three stages: (1) the early ‘concealed phase’, with non-apparent or subtle structural RV changes at which patients can already be at risk of SCD; (2) the ‘electrical phase’, characterised by T-wave inversions and terminal QRS prolongation on ECG, premature ventricular complexes (PVCs) and ventricular tachycardias (VT) with left bundle branch block morphology; (3) the ‘structural phase’ when structural modifications progressed into right or biventricular dilatation and potentially heart failure.13 Important differential diagnostic considerations (table 1) include idiopathic RV outflow tract (RVOT) VT, Brugada syndrome, myocarditis, sarcoidosis and non-ischaemic dilated cardiomyopathy (DCM).14 Differentiation can be challenging yet is crucial for appropriate clinical management.
Most common differential diagnostic considerations for ARVC
Pathophysiology
Structural changes
Focal structural myocardial lesions in ARVC typically manifest as fibrofatty replacement in the RV basal inferior wall, RV basal anterior wall and LV posterolateral wall, that is, the ‘triangle of dysplasia’.15 This is the result of progressive cardiomyocyte loss, starting in the subepicardial layer extending towards the endocardium leading to transmural thinning lesions.8 Although the exact molecular pathophysiology remains unclear, several hypotheses have been proposed.5 Most commonly, cardiomyocyte loss and fibrofatty replacement in ARVC are thought to be due to abnormal cell–cell adhesion with disruption of desmosomes and adherens junctions. This predisposes myocyte detachment and cell death, especially in combination with mechanical wall stress, for example, during exercise.
Arrhythmogenesis
In ARVC, monomorphic VTs most likely arise from fibrofatty lesions shaping highly arrhythmogenic re-entry circuits.16 However, as life-threatening arrhythmias can occur during the ‘concealed phase’ in the absence of (recognisable) structural heart disease, other mechanisms are likely involved as well. Recent preclinical studies revealed that loss of desmosomal integrity results in decreased gap junction protein (Connexin43) levels and sodium channel dysfunction, leading to abnormal impulse conduction.5 Furthermore, desmosomal mutations lead to dysregulated calcium handling, contributing to arrhythmogenesis in animal models.17 Concordantly, pathogenic variants in calcium handling protein genes (eg, PLN and RYR2) are found in some patients. Future research is required to further elucidate the pathological mechanisms underlying this early arrhythmic substrate.
Molecular genetics
Advances in molecular genetic research have led to the identification of various genetic substrates associated with ARVC. Most pathogenic variants are found in genes encoding the desmosome, predominantly PKP2 (table 2).18 The majority of variants have an autosomal dominant inheritance pattern with incomplete penetrance, with exceptions such as the fully penetrant TMEM43 p.S35L variant.19 Of note, some variants appear more frequent in LV-dominant phenotypes or DCM (eg, DSP, DSG2, DES, LMNA and PLN), and overlapping phenotypes are the rule rather than exception.20 Still, in approximately 30%–40% of index patients, no genetic substrate is found,21 indicating the role of other (epi)genetic, metabolic or even external causes for ARVC that have yet to be determined.
Genes associated with ARVC
Diagnosis
The 2010 TFC
No single test has sufficient sensitivity and specificity to serve as gold standard for ARVC diagnosis. Therefore, diagnosis is determined by a combination of clinical tests defined by a task force in 1994, the TFC, which was modified in 2010.2 Criteria considered to have high specificity (>90%) are classified as major and others as minor. The criteria are divided into six categories: (1) structure/function, (2) tissue characterisation, (3) repolarisation abnormalities, (4) depolarisation abnormalities, (5) arrhythmias and (6) family history. Per category, patients can fulfil only one minor or major criterion. At least two major, one major with two minor or four minor criteria are required for diagnosis (table 3). We will discuss some of the important new developments in ARVC diagnosis below.
The 2010 TFC for diagnosis of ARVC
Structure and function assessment
The 2010 TFC introduced quantitative echocardiography and cardiac MRI (CMR) criteria as alternatives to the previous standard of invasive angiography. While these criteria were recently shown to have high specificity (88%–99%), their sensitivity is relatively poor: 21%–29% for echocardiography and 46%–69% for CMR.22 23 A possible explanation for this limited sensitivity is that the primary condition for criteria fulfilment, detection of wall motion abnormalities, depends on subjective visual assessment. Besides being operator dependent, visual assessment may be insensitive for early signs of disease given the RV geometry and limited spatial resolution (particularly in echocardiography). Confirming this, echocardiography and CMR studies have shown objective assessment by deformation imaging (‘strain’) to be superior in detecting subtle motion abnormalities in early disease.13 24–26 Another limitation in the 2010 TFC may be the absence of multidetector CT (MDCT) as a useful alternative when obtaining CMR images is not possible due to implanted devices or claustrophobia.14
Tissue characterisation
Fibrofatty replacement is a typical sign of ARVC, and histological analysis has been a diagnostic tool for many years. Unfortunately, endomyocardial biopsy has a high rate of sampling error due to the segmental distribution of fibrofatty lesions.27 As the diagnostic yield is too low to justify the procedural complication risk, endomyocardial biopsy is usually reserved for cases in which mimics such as sarcoidosis cannot be otherwise excluded.
However, non-invasive detection of fat and fibrosis by CMR and MDCT is rapidly improving. Localised myocardial lesions may be detected by late gadolinium enhancement (LGE) CMR, with studies reporting sensitivities up to 88%.20 Alternatively, T1 mapping allows quantification of diffuse fibrosis and may detect ARVC preceding LGE, although the thin RV wall precludes T1 mapping analysis.28 Furthermore, contrast-enhanced MDCT low attenuation regions are also suggested to indicate fibrofatty infiltration.29 Although promising, future studies should determine if these techniques can differentiate ARVC from mimics. Advocating their use as diagnostic criterion seems premature as their true specificity for ARVC has yet to be determined.
Repolarisation abnormalities
The extent of precordial T-wave inversions (TWIs) on ECG in ARVC correlates to the degree of RV dilatation and is used for diagnosis.30 In addition to leads V1–3, indicating RV disease, the 2010 TFC includes TWI in V4–V6 as minor criterion, which may indicate LV involvement.31 As a result, this enables the inclusion of more LV-dominant cases in the TFC definition of ARVC, while in the future, this criterion may be more suitable as ALVC criterion.7
Depolarisation abnormalities
Depolarisation abnormalities in ARVC may manifest as prolonged terminal activation duration or epsilon waves on ECG or as late potentials on signal-averaged ECG (SAECG). Of these, SAECG and epsilon waves are currently under debate: SAECG had poor diagnostic performance in a recent validation study,22 and epsilon waves had high interobserver variability in an international expert panel.32 The latter is especially concerning considering its high impact as major criterion. Fortunately, the expert panel found that no patients depended on epsilon waves for their diagnosis, suggesting that it is a sign of advanced disease. As such, removing epsilon wave as diagnostic criterion will not affect ARVC diagnosis, while it may prevent harm caused by adjudication errors.
Arrhythmias
Both PVCs and VTs are included as diagnostic criteria for ARVC. While the PVC criterion depends on 24-hour count without requirements on morphology, strict morphological criteria apply for VT. In doing so, the Task Force aimed to avoid the overlap with idiopathic RVOT tachycardia. Since then, some authors have suggested that similar morphological criteria for PVCs would improve ARVC diagnosis.33 However, the feasibility of reliable morphology detection during ambulant Holter monitoring remains to be investigated.
Family history and genetics
Since the 2010 TFC family history and genetics criteria, all first-degree relatives and pathogenic variant carriers fulfil a major criterion. While this reflects the strong familial inheritance pattern of ARVC, this ‘head start’ in relatives could lead to false-positive diagnoses especially in the context of the incomplete penetrance. Indeed, a recent study revealed that relatives who depend on family history for diagnosis have generally benign follow-up.34 It is remarkable that the TFC considers having a first-degree relative with ARVC of equal weight as a confirmed pathogenic variant, since family history indicates 50% probability of harbouring a genetic predisposition (assuming an autosomal dominant inheritance pattern), while a confirmed genetic variant confers 100% probability. Indeed, the family membership criterion had much lower diagnostic value than positive genetic testing in a recent validation study.22 Future studies should systematically evaluate the role of family history and genetics in ARVC diagnosis.
Proposal for new ARVC, ACM and ALVC criteria: the Padua criteria
While the above outlined limitations of the 2010 TFC are widely recognised,35 changing the diagnostic criteria requires a strong evidence base as any change has major consequences for clinical practice and research.
A first step has recently been taken by Corrado et al,7 proposing new criteria for ARVC, ALVC and ACM: the Padua criteria. This proposal defines ACM as ‘a genetic heart muscle disease involving the RV, LV, or both, characterized by fibrofatty replacement predisposing to global and/or regional dysfunction, and ventricular arrhythmias independent of the ventricular dysfunction’. In this framework, ACM is subdivided as ARVC, ALVC or biventricular, with separate criteria for each entity. For ARVC, the main changes to the 2010 TFC include: wall motion abnormalities directly qualifying as minor criterion and transmural CMR LGE as major criterion. In addition, the Padua criteria remove SAECG and apply VT morphology criteria to the PVC criterion. As suggested by the authors, we emphasise that the Padua criteria should be evaluated in clinical validation studies prior to their clinical implementation.
Prognosis
Patients with ARVC have an average risk of 10%/year to develop ventricular arrhythmias including SCD.36 Of note, the only effective treatment to prevent SCD is the placement of an implantable cardioverter-defibrillator (ICD), which is invasive, has inherent complication risk and can impose physical and psychological burden to patients. As ARVC patients are often young, these burdens affect a significant part of their lives. Careful consideration of ICD indications is therefore warranted. However, the heterogeneity of SCD risk complicates decision making for ICD implantation. We will discuss the recent developments aimed at addressing this issue.
Expert statements and guidelines
Although many studies identified risk factors for arrhythmic events, translation to absolute risks relevant for clinical practice was lacking. Several expert consensus documents consolidated the available evidence in flow diagram algorithms to recommend ICD placement. Today, three algorithms are available: the 2015 international task force consensus (ITFC) statement on management of ARVC,16 the 2017 American Heart Association/American College of Cardiology/ Heart Rhythm Society guideline for management of ventricular arrhythmias37 and the 2019 HRS consensus statement on evaluation, risk stratification and management of ACM (figure 2).10 Of note, all three algorithms are based on expert opinion, and clinical validation studies to estimate their accuracy are lacking. Moreover, the algorithms do not account for incremental or interactive effects of multiple risk factors, which may limit their real-life accuracy.

Expert statement/guideline ICD indication algorithms. Overview of the three flow diagram algorithms for implantable cardioverter-defibrillator (ICD) indication, from the 2015 ARVC international task force consensus (ITFC 2015),16 the 2017 American Heart Association/American College of Cardiology/Heart Rhythm Society ventricular arrhythmia guideline (AHA/ACC/HRS 2017)37 and the 2019 arrhythmogenic cardiomyopathy Heart Rhythm Society consensus (HRS 2019).10 ARVC, arrhythmogenic right ventricular cardiomyopathy; NYHA, New York Heart Association; LVEF, left ventricular ejection fraction; PVC, premature ventricular complex; RA, right atrium; RVEF, right ventricular ejection fraction; VF, ventricular fibrillation; VT, ventricular tachycardia.
ARVC ‘risk calculator’
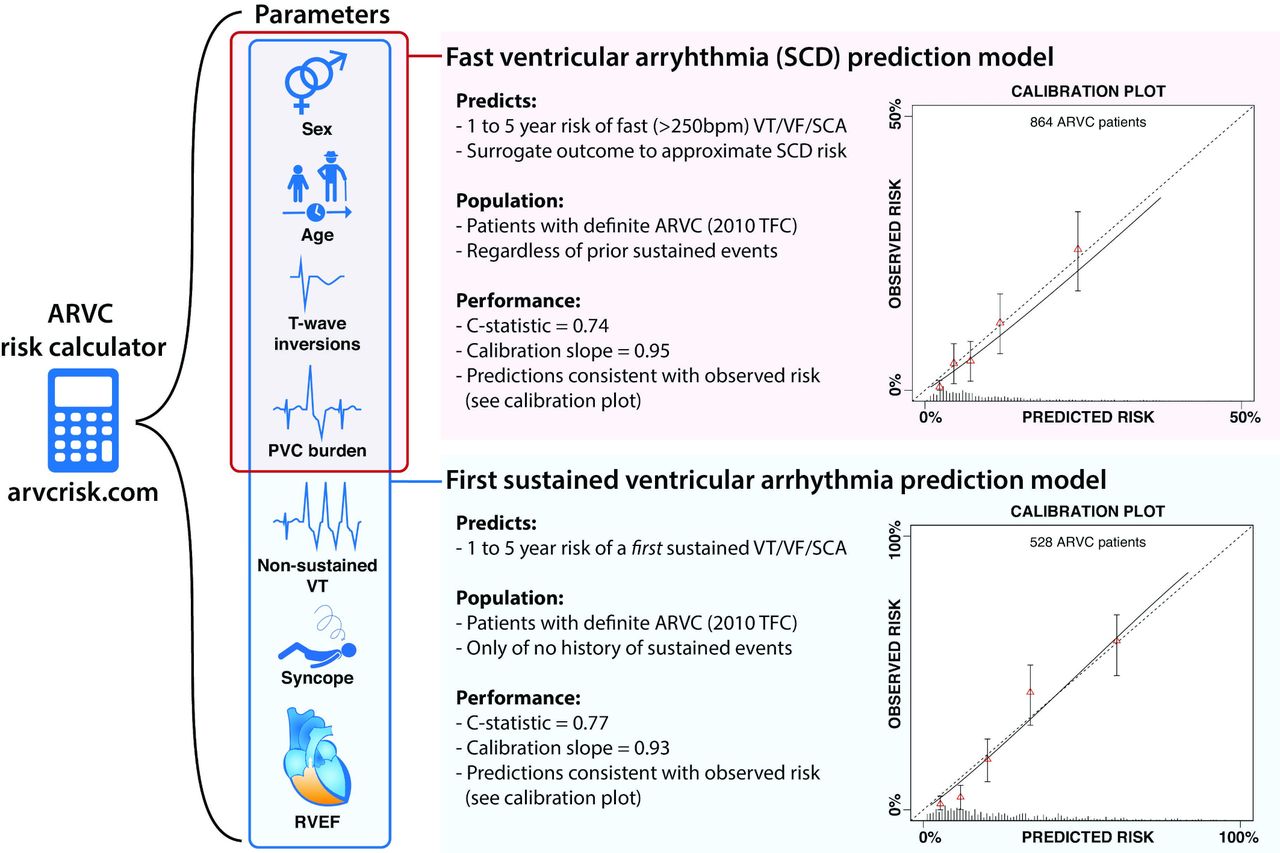
Traditional well-accepted univariable risk factors of arrhythmia in ARVC include prior VTs, right ventricular ejection fraction and left ventricular ejection fraction. However, multivariable models provide more accurate and quantitative estimations of arrhythmic risk. Two such models were recently developed in a large international cohort of ARVC patients: one to predict the first sustained ventricular arrhythmia in those without prior sustained events,38 and one to predict fast (>250 bpm) VT, ventricular fibrillation or sudden cardiac arrest/death (as SCD surrogate) in all patients.39 Both models are available as ‘risk calculator’ at www.arvcrisk.com (figure 3). As of today, four studies tested the calculator’s accuracy. First, Aquaro et al 40 showed that the calculator outperformed both the 2015 international task force and 2019 HRS algorithms in a cohort of 140 ARVC patients. Furthermore, studies by Aquaro et al 41 and Casella et al 42 confirmed excellent results in ARVC patients but reported that arrhythmic risk was underestimated in non-classical subtypes with LV involvement, suggesting this as a limitation. Interestingly, while underestimation was expected in athletes as exercise is not included in the risk calculator, Gasperetti et al 43 found accurate predictions in 25 athletes with ARVC. These results suggest a possible role for this risk calculator in clinical practice.

{kind=link}
{kind=link}
{kind=link}
ARVC risk calculator. The arrhythmogenic right ventricular cardiomyopathy (ARVC) risk calculator predicts ventricular arrhythmias in patients with ARVC by using two prediction models. One to predict the risk of fast (>250 bpm) ventricular tachycardia/fibrillation or sudden cardiac arrest (VT/VF/SCA) based on four risk parameters (red box), the other predicts the risk of any first sustained ventricular arrhythmia in those without a prior sustained event, using all seven risk parameters (blue box). The calibration plots of both prediction models are included (right side), plotting the predicted risk (X-axis) against the observed risk (Y-axis).38 39 PVC, premature ventricular complex; RVEF, right ventricular ejection fraction; SCD, sudden cardiac death; TFC, Task Force Criteria.
Clinical management
With no curative treatment options, clinical management is aimed at symptom reduction and prevention of disease progression and SCD. When an ICD is indicated as discussed above, both transvenous or subcutaneous are possible depending on preferences, vascular status and the need for pacing options such as bradycardia or antitachycardia pacing.37 Additional therapy options are discussed below.
Lifestyle
It is well appreciated that high-intensity or competitive exercise is associated with earlier disease onset, higher arrhythmic risk and structural disease progression in ARVC patients and at-risk relatives.44 45 As such, exercise restriction is strongly recommended for both patients and at-risk relatives. Unfortunately, it remains unclear to what extent exercise should be reduced to prevent harmful effects, while maintaining the physical and mental health benefits. The European Society of Cardiology guideline recommends a maximum of 150 min low-moderate intensity (3–6 metabolic equivalent) exercise per week in affected and at-risk subjects.46
Medication
Since arrhythmias in ARVC typically occur during exertion and are sensitive to ß-adrenergic stimulation,47 beta-blockers are recommended as first-line pharmacological agent. When unsuccessful, arrhythmic burden may be reduced using antiarrhythmic drugs, of which sotalol and amiodarone are considered most effective.48 Of note, none of these medications effectively reduce SCD risk. Pharmacological management of heart failure involves regular heart failure drugs, including beta-blockers, ACE inhibitors and mineralocorticoid inhibitors, but there are no ARVC-specific controlled trials confirming the effectiveness of this approach. Although ARVC-specific therapies are currently lacking, new therapeutic strategies targeting the Wnt/β and NFκB pathways show disease regression in animal models and may be promising in the future.49
Cardiac catheter ablation and transplantation
In patients with frequent monomorphic VT, radiofrequency catheter ablation can be considered for symptom relief, but full resolution of ventricular arrhythmias is virtually impossible due to the progressive nature of disease. In addition, since arrhythmic substrates in ARVC are predominantly located on the epicardium, an epicardial approach is usually necessary. Indeed, several studies have shown significantly better results with an epicardial compared with endocardial approach.50 In patients with untreatable ventricular arrhythmias or congestive heart failure refractory to therapy, cardiac transplantation can be considered as definitive solution.11
Conclusions
ARVC is an inherited cardiomyopathy with high risk of ventricular arrhythmias that may lead to SCD at young age. Accurate early detection of disease is essential for SCD prevention, which was significantly advanced by genetic testing identifying those at risk at preclinical stages. However, clinicians are challenged by incomplete penetrance and highly variable disease expression among individuals. To overcome these challenges, the recent years have witnessed research into solutions that tailor clinical care to an individual level. To improve early disease detection, recent studies showed promising results using non-invasive tissue characterisation and deformation imaging. To improve risk stratification, a multivariable prediction model for ventricular arrhythmias was developed. Furthermore, ARVC is now increasingly recognised as being part of a wider disease spectrum involving both ventricles: ACM. While uniform definitions are still lacking, subclassifying patients into similar, more uniform phenotypic groups may benefit future research and improve clinical management.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @laurensbosman
Contributors LPB and ASJMtR contributed equally.
Funding LPB is supported by a Netherlands Heart Institute Fellowship 2020. The Dutch ACM Registry is supported by the Dutch Heart Foundation (CVON eDETECT 2015–12, CVON2018- 30 PREDICT2 and a personal grant to ASJMtR (grant number 2015T058).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; internally peer reviewed.