Article Text

Abstract
Two new cases of dilated cardiomyopathy (DC) caused by dystrophinopathy are reported. One patient, a 24 year old man, had a family history of X linked DC, while the other, a 52 year old man, had sporadic disease. Each had abnormal dystrophin immunostaining in muscle or cardiac biopsy specimens, but neither had muscle weakness. Serum creatine kinase activity was raised only in the patient with familial disease. Analysis of dystrophin gene mutations showed a deletion of exons 48–49 in the patient with familial DC and of exons 49–51 in the other. Dystrophin transcription in cardiac tissue from the patient with sporadic disease showed abundant expression, predominantly of the muscle isoform. This study, together with previous reports, suggests that some patients with DC have a dystrophinopathy that can be diagnosed using a combination of biochemical and genetic analyses.
- dilated cardiomyopathy
- dystrophin
- Becker muscular dystrophy
Statistics from Altmetric.com
Duchenne’s (DMD) and Becker’s muscular dystrophies (BMD) are allelic X linked neuromuscular disorders. They result from mutations in the dystrophin gene that, when severe as in DMD (nonsense or out of frame mutations), lead to lack of expression of the sarcolemmal protein dystrophin.1 Up to one third of all cases of DMD and BMD arise from de novo mutations, so that a significant proportion of affected males have no family history of the condition.1 ,2 The absence of dystrophin in DMD causes severe and progressive muscle weakness, loss of ambulation before the age of 13, and death in the late teens or early twenties.3BMD is a milder form of muscular dystrophy in which individuals are ambulant after the age of 16. These individuals usually have in frame deletions and residual expression of a partially functional dystrophin in the muscle.3
Cardiac involvement is an integral part of DMD and BMD.4-7 In rare instances, however, patients can suffer from dilated cardiomyopathy (DC) as the only manifestation of a dystrophinopathy. This has been now recognised in families with typical X linked DC8-11 and in patients with sporadic disease.12-14 Unlike patients with DMD or BMD, these patients did not have symptoms of muscle weakness and the only sign of neuromuscular involvement was raised serum creatine kinase (CK) activity. Several patients had mutations clustered in the 5′ end of the gene9 ,11 ,12; a region not affected by mutations usually found in DMD and BMD. DC has also been reported in asymptomatic female carriers of DMD and BMD.15
Two cases of familial (X linked) and sporadic DC secondary to a dystrophinotrophy are reported here.
Patients
The two patients described in this study originate from Italy. Ethical approval was obtained from the committees of the two hospitals involved in their assessment. Informed consent was obtained from each patient.
PATIENT 1
The first patient, a 24 year old man, was admitted in August 1991 after the recent onset of dyspnoea with mild physical activity. DC was diagnosed (left ventricular end diastolic diameter 73 mm; left ventricular end diastolic volume 107 ml/mq; left ventricular ejection fraction 27%) after exclusion of active myocarditis by endomyocardial biopsy and coronary artery disease by angiography. An electrocardiogram showed Q waves in the inferior and posterior leads and incomplete right bundle brunch block. Sustained ventricular arrhythmias were detected after 24 hour Holter monitoring. Among the biochemical investigations, constantly raised serum CK activity (MM isoform, ranging between 540 and 867 U/l, normal < 200 U/l) was found. Neurological investigation failed to show any weakness or muscle wasting or hypertrophy. The patient denied symptoms of neuromuscular involvement such as cramps or myalgias associated with exercise. He improved following treatment with digitalis, enalapril, and metoprolol and remained in New York Heart Assocation (NYHA) class I–II. During follow up the left ventricular dimension slightly diminished (left ventricular end diastolic diameter 70 mm), although the systolic function remained significantly depressed (left ventricular ejection fraction 37%) with diffuse wall motion abnormalities, more evident in the inferoposterior region. Mild right ventricular dysfunction was observed at radionuclide angiography (right ventricular ejection fraction 35%). At the last follow up (1996) the patient was in NYHA class II with a peak consumption of oxygen of 14.4 ml/kg/min (anaerobic threshold 5.8 ml/kg/min) and without evidence of electrical ventricular instability with Holter monitoring or during exercise.
Analysis of the pedigree disclosed a strong family history of DC suggestive of an X linked inheritance (fig 1). Individuals II:3, III:6, and III:11 (fig 1) had been affected by DC and died at the ages of 36, 42, and 38, respectively. Moreover, a maternal aunt (individual II:5, fig 1), an obligate carrier for the condition, also died of DC at the age of 57. Interestingly, individual III:8 had DMD and died because of respiratory complications at the age of 22. All other affected members did not have signs of skeletal muscle involvement, at least as far as could be assessed from their medical records and questioning their relatives. A needle muscle biopsy specimen from the propositus (individual III:4, fig 1) was obtained to investigate the possibility of an underlying dystrophinopathy.
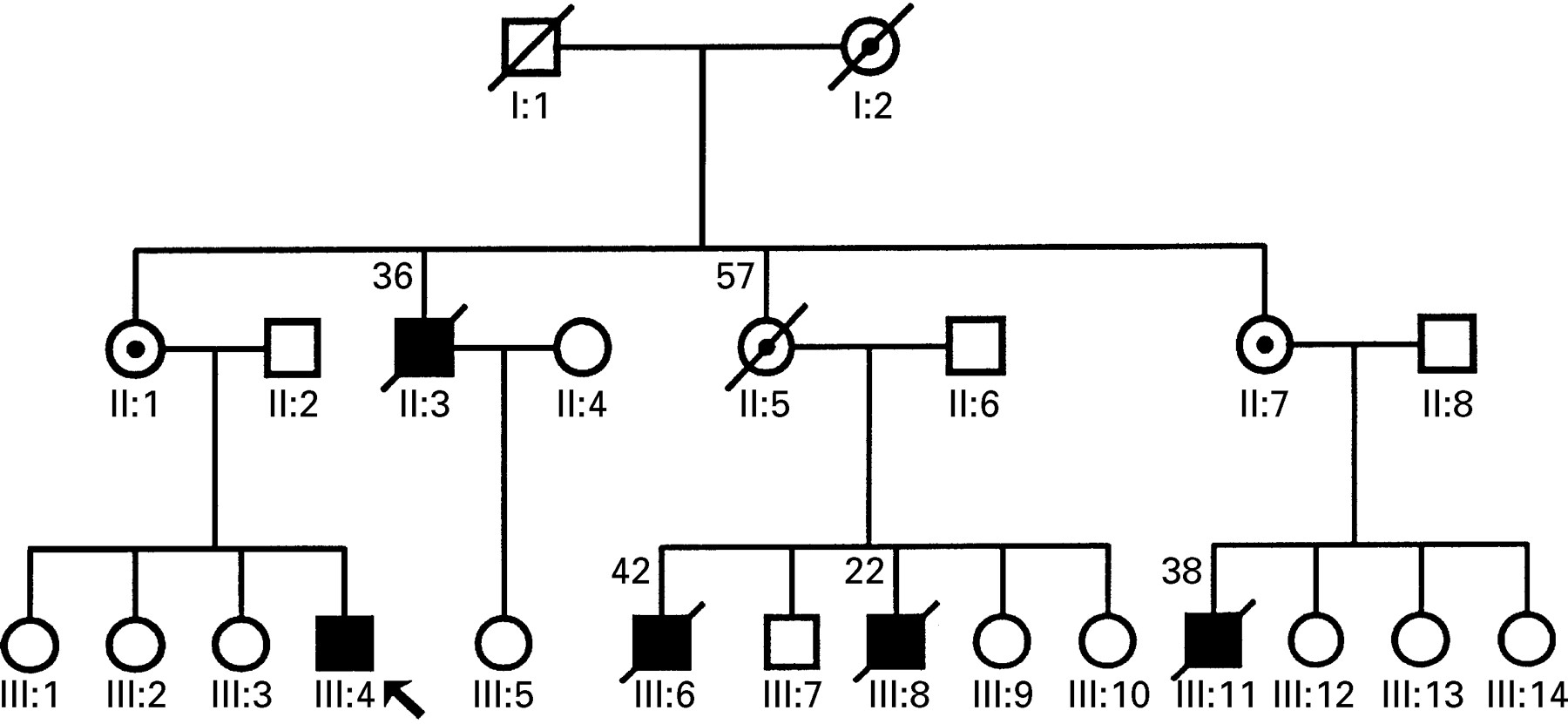
Pedigree of family with X linked cardiomyopathy. Open symbols, unaffected individuals; closed circles, affected individuals; barred symbols, deceased (number above is age at death); dotted circles, obligate carriers; arrow, propositus.
PATIENT 2
The second patient, a 52 year old man, died shortly after cardiac transplantation in 1994. He was employed in the chemical industry and was physically active until the age of 50 when he presented with the first symptoms of cardiac failure. There was no family history of cardiovascular disease. The patient smoked 20 cigarettes daily and admitted moderate alcohol intake until the age of 50, when he developed a gastric peptic ulcer that eventually required surgical excision. On examination, aged 52, he was dyspnoeic at rest and a systolic murmur was present over the mitral valve. There was an additional third tone, but no raised jugular venous pressure or peripheral oedema. His liver was enlarged and neurological examination failed to show muscle atrophy or pseudohypertrophy. His muscle strength was normal, as was serum CK activity (84 U/l). Negative T waves in leads V5–V6 were seen on the electrocardiogram. Echocardiography showed a dilated left ventricle (left ventricular end diastolic diameter 70 mm, left ventricular ejection fraction 20%). The right ventricle and both atria were also dilated. Moderate mitral and tricuspidal valve regurgitation was also found. Right heart catheterisation disclosed pulmonary hypertension, (pulmonary artery pressure 68/30 mm Hg, mean 42 mm Hg), and a reduced cardiac index (2.0 l/min/m2). Despite medical treatment with angiotensin converting enzyme inhibitors, digitalis, and diuretics the patient deteriorated clinically and he underwent cardiac transplantation nine months later. In the early postoperative period he suffered from multiorgan failure and died shortly after.
Methods
IMMUNOHISTOCHEMICAL STUDY OF SKELETAL AND CARDIAC MUSCLE
A cardiac biopsy specimen obtained at cardiac transplantation, and a needle biopsy specimen of skeletal muscle taken from patient 1 were immediately frozen in liquid nitrogen cooled isopentane and stored at −70ºC or in liquid nitrogen. The specimens were fully processed for histological, histochemical, and immunohistochemical examination.16 In particular, unfixed cryostat sections (6 μm) were immunostained using a panel of six antidystrophin antibodies, including the monoclonal antibody DYS1808 (Ylem, Italy), as already described.17
DNA STUDIES
DNA was isolated from leucocytes by standard methods. Multiplex DNA amplification of dystrophin exons was carried out according to previously described techniques.18 ,19
REVERSE TRANSCRIPTION AND POLYMERASE CHAIN REACTION
Total RNA20 was isolated from control frozen skeletal muscle and heart and from the left ventricle of patient 2. cDNA synthesis was performed using random hexanucleotide primers, following the procedure already described.21 ,22Polymerase chain reaction was performed using forward primers designed to amplify the muscle, brain, and Purkinje cell isoforms and an exon 6 reverse primer as already described.21
Results
IMMUNOHISTOCHEMICAL ANALYSIS
The skeletal muscle biopsy specimen from the propositus showed mild dystrophic changes, characterised by an increase in internal nuclei (12%), mild variability of fibre size with hypotrophic and hypertrophic fibres, and rare fibre splittings in an otherwise well preserved muscle. There were no signs of degeneration or regeneration and only a marginal increase in connective tissue. Immunohistochemistry performed with the panel of antidystrophin antibodies showed variability in the immunoreaction between adjacent fibres (fig 2), the level of dystrophin expression being weaker than control muscle (fig 2).
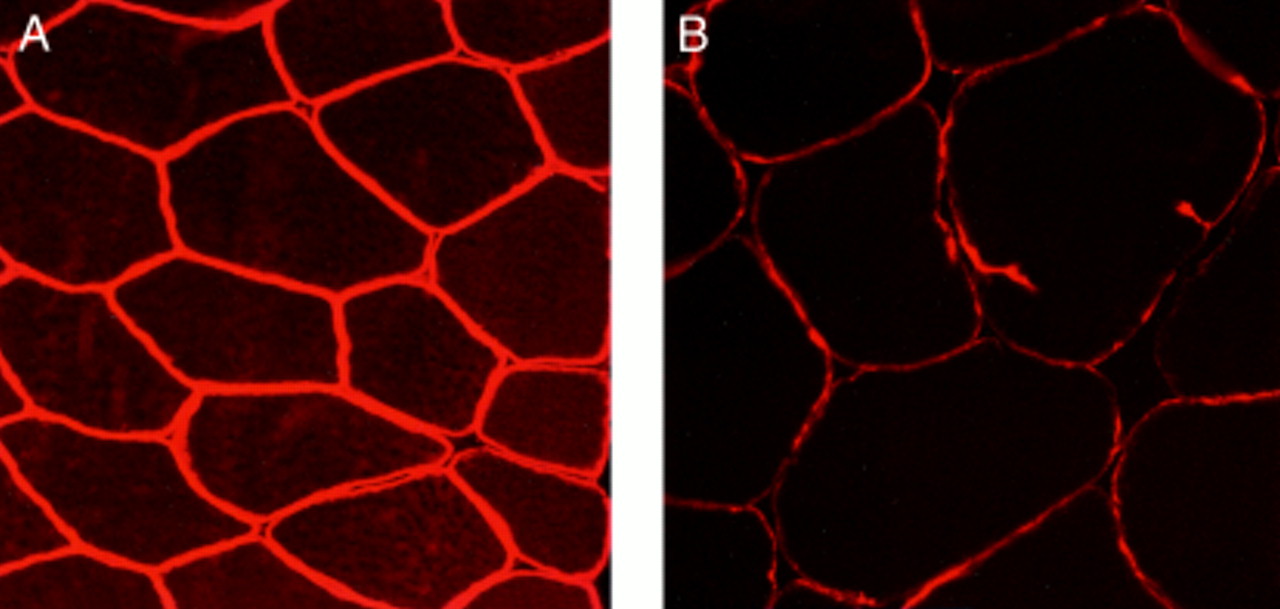
Immunohistochemical staining of skeletal muscle with Dys-III N terminus antidystrophin antibodies. (A) Normal muscle: all fibres equally and intensely stained (original magnification ×280). (B) Muscle from patient 1: staining is weaker than control muscle, suggesting a dystrophinopathy (original magnification ×280).
Skeletal muscle samples were not available from patient 2. His cardiac biopsy specimen showed significant fibrosis, separating individual cardiomyocytes in some areas, and gross variability of fibre size mainly due to hypertrophic cardiomyocytes. A few cardiac cells undergoing degeneration were also seen. Immunostaining with antibodies directed towards the N terminal, mid-rod, and C terminal domain of dystrophin was strong and continuous, although there was some variability between adjacent fibres, mostly visible using N terminal antibodies (fig 3). There was no staining, however, using the terminal rod domain antibodies DYS-1808, suggesting the presence of a mutation in the genomic region encoding for this epitope (fig 3). Previous studies indicate that such region exists between exons 49 and 51.23

Top, immunohistochemical staining of cardiac muscle from patient 2 using antidystrophin antibodies. Bottom, protein dystrophin and epitopes of three of the antibodies used in the study. Top left, N terminus antidystrophin antibodies (Dys3) with a near normal reaction; top middle, no staining was visible with mid-rod domain 1808 antidystrophin antibodies; top right, reduced staining with Dys2 C terminus antidystrophin antibodies (original magnifications ×320).
Western blot analysis22 showed a slight reduction in the amount of dystrophin expression with a small decrease in molecular weight (fig 4).

Western blot analysis of cardiac biopsy from (A) control and (B) patient 2, probed with mid-rod antidystrophin antibody (P6).22 Note the slight reduction in abundance and molecular weight of dystrophin in the patient (arrow).
DYSTROPHIN GENE ANALYSIS
Multiplex ploymerase chain reaction deletion study showed deletion of the central region of dystrophin (data not shown). In the patient with sporadic disease in frame deletion of exons 49–51 was found, while the propositus of the family with DC had a deletion of exons 48–49, which was subsequently confirmed in the obligate carriers (II:1 and II:7).
TRANSCRIPTION STUDIES
Transcription of dystrophin muscle, brain, and Purkinje cell isoforms from the heart of patient 2 resulted in a pattern of isoform expression indistinguishable from that in control heart. Expression of the muscle isoform was high, while that of the brain was low. Purkinje cell isoforms were not expressed (fig 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Amplification of cDNA from normal cardiac muscle (C) and the cardiac biopsy from patient 1 (252). Dystrophin muscle isoform (M) was amplified as a 494 base pair fragment, the brain isoform (B) as 482 base pairs, and the Purkinje cell isoform (P) as 503 base pairs. The cardiac muscle biopsy from patient 1 showed transcription of trace amount of isoform B and higher expression of isoform M, but no expression of isoform P. The control cardiac tissue showed an identical pattern of isoform transcription with a trace amount of isoform B and high expression of isoform M. Lane K, molecular weight marker.
Discussion
Cardiac involvement is very common in DMD and BMD. Almost all patients with DMD have signs of cardiac involvement in the late stages of their disease,5 while various authors have reported an incidence of cardiac involvement in patients with BMD of approximately 60–65%.5-7
Our group recently reported that the dystrophin gene is also involved in some patients with X linked DC.9 ,11 Dystrophin expression and transcription in these families showed that dystrophin was absent in the heart but not in skeletal muscle, thereby providing a biochemical explanation for severe cardiomyopathy.11 21 24 The fact that several patients with X linked DC carried unusual but similar mutations in the extreme 5′ end of the gene9 ,11 ,12 suggests a link between these mutations and the prevalent cardiac involvement. In contrast, there is still no information on the precise prevalence of dystrophin abnormalities in a population of patients with DC. The only study that has addressed this issue was performed on 27 males with DC, but dystrophin deletion was not found, suggesting that the frequency of dystrophinopathy in the population of patients with DC is probably low.25
Two further cases of DC without muscle weakness due to a dystrophinopathy are reported here. In each patient there was a deletion in the central rod domain of dystrophin, a region located in the dystrophin deletion “hot spot”. Identical mutations are typically associated with mild BMD.2-26 The reason for the lack of skeletal muscle involvement in our patients with a “typical BMD deletion” is unclear, but intriguing, especially considering that the patient with sporadic disease had normal serum CK activity. The mechanism of cardiac involvement in our patients is likely to be different from that in patients with mutations in the 5′ of the gene, in whom cardiac expression of dystrophin could not be found.11 ,24 Indeed, high levels of cardiac dystrophin expression were seen in the patient with sporadic disease with a deletion of exons 49–51 using both immunocytochemistry and western blot analysis. Moreover, the transcription pattern in this patient was indistinguishable from that seen in normal heart—that is, prevalent expression of the muscle isoform with lower expression of the brain isoform.21 Unfortunately, cardiac muscle was not available from the other patient with a deletion of exons 48–49.
Various authors have reported a high incidence of cardiac involvement in patients with BMD with deletions involving exon 49, while deletion of only exon 48 are less frequently associated with a cardiomyopathy.7 ,27 ,28 It has been proposed that intron 48 might contain sequences relevant to the function of dystrophin in cardiac muscle. This intronic sequence would be removed or preserved by an intragenic deletion involving exon 48, depending on the break point within intron 48, while any deletion encompassing both exons 48 and 49 would remove these sequences.7 ,28 Another possibility is that exon 49 contains a domain with an essential function for the heart. Recent analysis of dystrophin has shown, for example, the presence of a calcium binding domain within the cysteine rich region,29 while a WW domain, involved in mediating protein protein interaction, is present in exons 62–64.30 Exon 49 is located in the central region of dystrophin, however, where the protein has an organisation similar to that of the coiled coil repeats of spectrin.31 Exon 49 is contained entirely in repeat 1931 and therefore it seems unlikely to have a structure that might account for a specific and unique function in the heart.
We have no explanation as to why a cousin of the patient in the family with DC was affected by DMD, while the three remaining individuals had no signs of skeletal muscle involvement. One possibility is that of a second mutation in the dystrophin gene. Such an eventuality has been reported,32 ,33 although it is rare. Unfortunately, DNA or skeletal muscle was not available from this patient.
Our results suggest that a deletion of the dystrophin region involved in the cases reported here can give rise not only to BMD, but also to DC. Intriguingly, serum CK activities can be normal, as highlighted by our patient with sporadic disease. High serum CK activities are therefore not essential for suspecting DC secondary to a dystrophinopathy.
In conclusion, two cases of DC secondary to a dystrophin mutation are reported. The findings suggest that involvement of dystrophin can be responsible for idiopathic DC, although its incidence is probably low.25 Combined genetic and biochemical studies on a larger patient population are needed to clarify the prevalence of dystrophin abnormalities in patients with DC.
Acknowledgments
This work was financed by grants from the Muscular Dystrophy Group of Great Britain and Northern Ireland, the British Heart Foundation (FM), the Associazione Amici del Cuore of Trieste, the National Research Council (CNR 95, 04378.CT04, 95.00824.CT04, 95.01670.CT04, A195.00346.04), and Telethon-Italy (E.291; CC).