Article Text

Abstract
OBJECTIVE To predict the growth in demand for long term follow up of adults with congenital heart disease.
DESIGN Observed diagnoses of congenital heart disease in infancy and childhood were adjusted for observed infant survival, predicted further survival to age 16 years, underascertainment in older childhood, and predicted need for long term follow up.
SETTING The resident population of one health region in the UK.
PATIENTS All confirmed cardiovascular malformations diagnosed in 1985 to 1999 in children born in 1985 to 1994.
RESULTS 1942 cases of congenital heart disease were diagnosed in infancy in a population of 377 310 live births (5.2/1000). 1588 (82%) survived to 1 year and 1514 were predicted to survive to age 16. 605 further diagnoses were made in childhood—678 when adjusted for underascertainment. Thus, 2192 children were predicted to reach age 16, of whom 784 would require long term follow up in adult life. The adult population would comprise 28% complex, 54% significant, and 18% minor congenital heart disease. These figures predict the need for adult follow up of congenital heart disease of over 200 extra cases per 100 000 live births each year or over 1600 extra cases a year every year in the UK.
CONCLUSIONS The need for follow up of congenital heart disease in adult life is likely to grow linearly, with increasing complexity and increasing need for reinvestigation and reintervention with time. Appropriate provision should be made for adequate manpower, resources, and facilities for care of these patients.
- adult congenital heart disease
- resources
- patient survival
Statistics from Altmetric.com
Improvements in diagnosis, medical treatment, and surgical repair are changing the pattern of survival of congenital heart disease. In recent years the survival of more complex types of congenital heart disease has improved, leading to an increasing work load for follow up, reinvestigation, and reoperation. Over the same time, the care of adults with congenital heart disease has begun to develop as a separate subspecialty.1 It seems likely that all but those with the simplest forms of congenital heart disease will require specialist follow up, and this will be the main growth area in care of patients with congenital heart disease.2 So far there has been inadequate planning for the future care of such patients and there are no population based data to assess the future growth of this subspecialty.2 ,3 The aim of this study was, therefore, to predict the likely future growth of the population of survivors of congenital heart disease requiring follow up beyond the age of 16 years.
Methods
POPULATION
The former Northern Health Region comprises the counties of Cumbria, Northumberland, Tyne & Wear, Durham, and Cleveland and has a resident population of just over three million. All infants and children with suspected heart disease from 15 of the 16 health districts are referred to a single centralised paediatric cardiology centre.4 Children from the health district of South Cumbria are referred elsewhere for geographical reasons and so were not included in this study.
DATA SOURCES
The paediatric cardiology database contains details of all infants and children with structural congenital heart disease born since 1 January 1985.4 Ascertainment was prospective for all diagnoses made from 1990 onwards and retrospective for diagnoses made between 1985 and 1989. Ascertainment is thought to be complete for all significant and complex heart disease with some underascertainment of minor malformations in infancy in the early part of the study.5
Diagnoses made in infancy were limited to babies born to mothers resident in the region at the time of birth. There was no significant inflow or outflow of population within the first 12 months.6 Diagnoses made after the first birthday were limited to those made in children resident in the region at the time of diagnosis. Infants and children moving into the region and referred for follow up of congenital heart disease that had already been recognised were not included. This allows us to compensate for immigration and emigration to other regions. National statistics show no significant net inflow or outflow of population during the period of the study.6 The Office of National Statistics provided data on the regional birth rate.
CASE DEFINITION
Cardiovascular malformations were defined according to the generally accepted definition of “a gross structural abnormality of the heart or intrathoracic great vessels that is actually or potentially of functional importance”.7 In keeping with previous population studies, children with isolated cardiac arrhythmias, cardiomyopathy, acquired heart disease, isolated bicuspid aortic valve, mitral valve prolapse without regurgitation, isolated dextrocardia, cardiac tumours, patent ductus associated with prematurity, and atrial septal defect undergoing spontaneous closure in infancy were excluded. All cases were classified according to the main anatomical diagnosis. Children with more than one cardiac malformation received a single diagnosis based on a previously described anatomical hierarchy.5 Diagnoses classified as “miscellaneous” in tables 1 and 2 mainly included tricuspid valve abnormalities, pulmonary artery branch stenosis, great artery and coronary artery malformations, and left ventricular outflow obstruction. As described previously each anatomical diagnosis was also classified as complex, significant or minor.5 Complex malformations were characterised by an absent, hypoplastic or common valve or chamber (including congenitally corrected transposition of the great arteries and complete atrioventricular septal defect); significant malformations involved hearts that had four valves and chambers but would require intervention; and minor defects were mainly smaller ventricular septal defects and less severe aortic or pulmonary valve stenosis. Patients with ventricular septal defect, aortic valve stenosis or pulmonary valve stenosis were divided into those dying or undergoing surgical repair in infancy (significant) and those alive and unoperated by one year or diagnosed after infancy (minor).5 Late presentation of a large ventricular septal defect, inoperable because of pulmonary vascular disease, was not encountered in this study but would have been classified as significant.
Observed survival in infancy, predicted survival throughout childhood, and predicted requirement for follow up in adult life
Observed cases, predicted survivors, and predicted need for follow up in adult life
MODEL DESIGN
Because the database contains data for patients live born since January 1985 the follow up ranged from a minimum of one year for babies born in 1998 to a minimum of 13 years for babies born in 1985.5 The study was based on a theoretical population born in 1985 to 1994 with 10 years' ascertainment for all ages from birth to the 16th birthday (fig 1). These years were chosen because the oldest patients are about to graduate to the adult congenital heart clinic. They provide sufficient data for study, yet minimise the need for adjustment for underascertainment of diagnoses at older age. We have 100% ascertainment for the first five years of life and then diminishing ascertainment from 90% for 5 year olds to 10% for 13 year olds. To predict the size of the theoretical population, extrapolation from the observed population is required (fig 1) and assumes constant underascertainment. The required “correction factor” ranges from 1.1 for 5 year old children to 10.0 for 13 year old children to predict the situation if full ascertainment for 10 years at each age were achieved. The number of cases of each diagnosis identified at each age was determined from the database. Each age cohort was then corrected for underascertainment of each diagnosis (using the correction factor detailed above) and the total predicted numbers of each diagnosis at each age were then calculated.

Data available on all diagnoses in babies and children born in 1985 to 1994. Ascertainment varies from 13 years for births in 1985 to four years for births in 1994. Data range from 10 years for 0–4 year olds to one year for 13 year olds. The figure shows data available (hatched) and data extrapolated (grey). The effect of the adjustment is shown in fig 2.
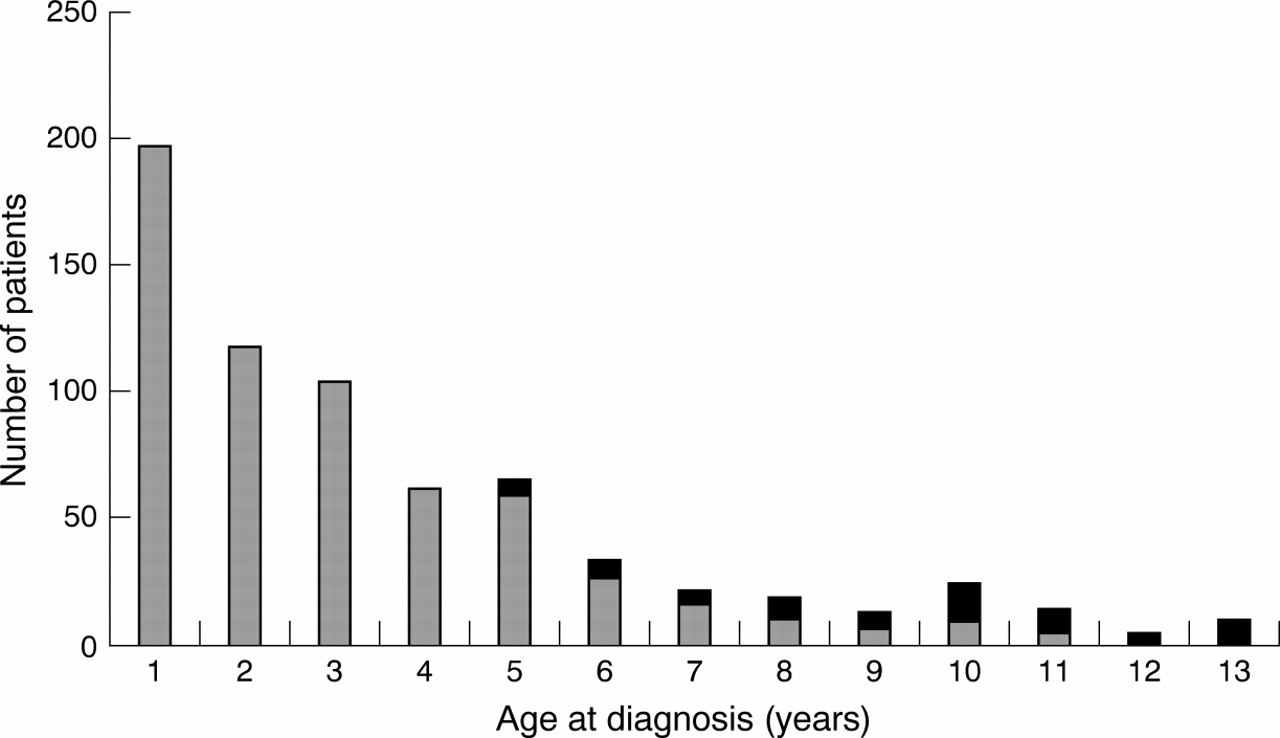
There is potentially an increasing error with older age at diagnosis because of the increase in the correction factor required but, in fact, the number of diagnoses diminishes rapidly with increasing age. Figure2 shows that the very large majority of diagnoses are made early in childhood so the potential error from extrapolation later in childhood is much reduced. Even with correction for underascertainment there are very few diagnoses over the age of 10 years (fig 2). Because of the design of the database we have no diagnoses in children aged 14 years and older. The likely error from excluding all children diagnosed at the age of 14 and 15 years is very small.

The effect of adjustment for underascertainment. All diagnoses at 1–13 years are shown (grey bars) with the effect of extrapolation to compensate for underascertainment at ages 5–13 years (solid bars). The difference between raw and adjusted figures is very small and is shown for individual diagnoses in table 2.
The study identified all live born newborns with heart disease during infancy and documented survivors at the end of the first year. The likely survival for each patient diagnosed from the age of 1 year to 16 years was predicted from the average of up to three published reports per diagnosis, which gave survival data as shown in table1.8-54 This gave a predicted theoretical population of 16 year old survivors of heart disease presenting in infancy. New diagnoses made at 1–13 years (corrected for underascertainment for 5–13 year olds as described above) produced a second theoretical childhood population, which was added to the infant survivors (table2). It was assumed that all those diagnosed beyond infancy would survive until the age of 16 years. This produced a total number of patients reaching the age of 16 alive and a description of the spectrum of congenital heart disease they would have. Reports in the literature were used to predict the need for specialist follow up into adult life (table 1). This ranged from 100% for most serious forms of heart disease to 0% for patent ductus that was successfully closed or occluded in childhood. We have assumed, for instance, that 10% of children with ventricular septal defect would require continued follow up beyond 16 years of age, either for their ventricular septal defect or for associated aortic valve regurgitation, mitral valve regurgitation, repaired coarctation, subaortic stenosis, residual patch leak, permanent pacemaker, etc.55 This might be considered to be either an under- or overestimate and could be adjusted accordingly. We could thus deduce the number of patients requiring follow up and from this the annual number for our population. Knowing the relation between our regional birth rate and national figures, we could also predict the requirements for the UK.56
Results
During the 10 year study period there were 377 310 live births in the population. The birth rate diminished from 38 865 in 1985 to 35 026 in 1994.
In the same population 1942 cases of congenital heart disease were diagnosed in infancy (5.2/1000) with 1588 (82%) still alive at the age of 1 year. Observed total infant survival varied from 0% for hypoplastic left heart to 100% for small ventricular septal defect, mild pulmonary stenosis, and mild aortic stenosis. The spectrum of congenital heart disease at birth and at the age of 1 year is shown in table 2.
The number of new diagnoses of congenital heart disease made after infancy diminished with age. Even after correction for underascertainment in later years (as described above and as shown in fig 2) it is still apparent that most childhood diagnoses are made before school age. As can be seen from fig 2, 74% of all cases are diagnosed in infancy, 18% before school age (age 1–4 years) and 8% in school age (age 5–13 years). The most common abnormalities diagnosed after infancy were ventricular septal defect, patent ductus arteriosus, atrial septal defect, and pulmonary valve stenosis (table2), but there were also significant numbers of important malformations such as coarctation of the aorta and a few cases of more complex heart disease such as pulmonary atresia and tetralogy of Fallot.
Combining data for the survivors of heart disease diagnosed in infancy predicted to survive another 15 years with new diagnoses in childhood produces the spectrum of congenital heart disease in children reaching the age of 16 years (table 2). The predicted need for follow up into adult life ranged from 0% for closed patent ductus arteriosus to 100% for all major heart defects, and produced the predicted spectrum of heart disease requiring long term follow up beyond 16 years of age (table 2).2 As can be seen, the main diagnoses were repaired complete atrioventricular septal defect, atrial repair of transposition of the great arteries, repaired tetralogy of Fallot, repaired coarctation, and ventricular septal defect with significant numbers of mitral regurgitation, aortic stenosis, etc.
The birth rate in our catchment area comprises 4.86% of total births in the UK. The final column in table 2 predicts the total annual increment in patients requiring follow up of congenital heart disease into adult life in the UK.
Discussion
This study found that 74% of all congenital heart disease recognised in childhood is diagnosed in infancy. In 1985 to 1994 there was a total mortality in infancy of 18% but the predicted survival for a further 15 years for those surviving infancy is 96%. A further 18% of heart defects were diagnosed beyond infancy but before school age (1–4 years) so that 92% of all cases were recognised by the age of 4 years. Another 8% of children with cardiovascular malformations were first diagnosed during school age (5–13 years). Our study predicts the need for follow up beyond 16 years of age for repaired or unrepaired congenital heart disease in the UK of over 1600 cases extra each year or over 200 cases per 100 000 live births.
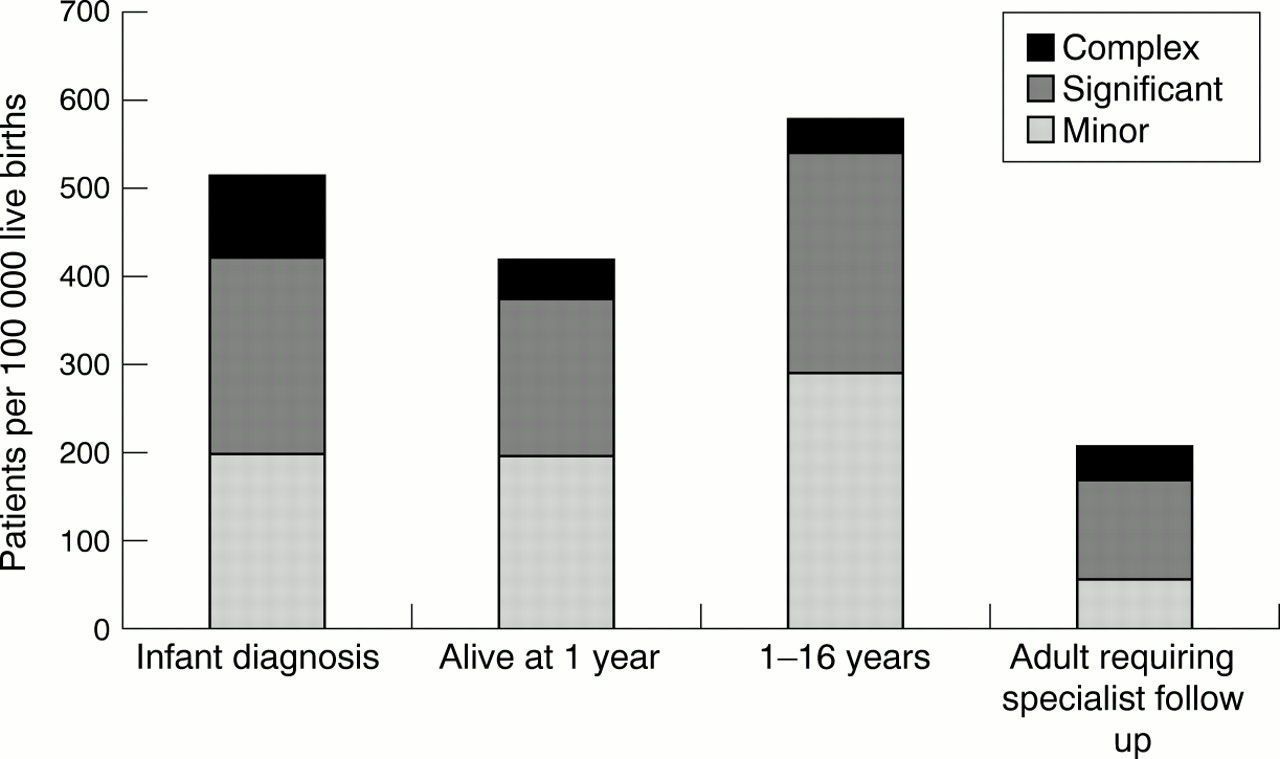
The spectrum of complexity of congenital heart disease diminishes throughout childhood—partly as a result of higher early mortality caused by serious malformations and partly because of later diagnosis of more minor abnormalities—but increases in adult life because many lesser defects do not require follow up (figure 3).

{kind=link}
{kind=link}
{kind=link}
The spectrum of complexity of congenital heart disease at different stages of follow up. Complexity decreases during childhood but increases in adult life if children with minor malformations that do not require long term follow up are excluded.
Numerically, the more complex malformations, including those suitable only for single ventricle repair (such as mitral atresia, tricuspid atresia, and double inlet ventricle) and others such as pulmonary atresia with ventricular septal defect or truncus arteriosus, are less common but they require more intensive follow up and will need more reinvestigation and repeat intervention.
We have restricted our analysis to structural heart disease but we recognise that children with cardiac tumours, Marfan's syndrome, cardiomyopathy, primary arrhythmia, and various other cardiac problems also require follow up. Thus we have underestimated the total number of patients graduating from the paediatric clinic to the adult congenital cardiology clinic. We also recognise that survival is constantly improving with advances in earlier diagnosis, surgery, and postoperative care so that the numbers of patients requiring follow up will continue to increase and the complexity of their cardiac problems will also increase.
In the very long term it is possible that a policy of antenatal diagnosis of cardiovascular malformation and termination of pregnancy will reduce the number of live born babies with complex malformations. The overall effect of this is likely to be very small57and would take many years to have a significant influence on the number of survivors in adult life. Such an effect on prevalence at live birth has so far not been demonstrated in our own region5 or in other studies.58
Most congenital heart disease is now survivable and late deaths in childhood or early adult life are relatively few.35 ,59 A review of adults with congenital heart disease followed up in Toronto, Canada showed an annual mortality rate of 13.9/1000 patient years (1.4%) so we can predict almost linear growth in the patient population in the foreseeable future.60
Few previous reports have considered the implications of this increasing workload. Gatzoulis and colleagues61 reported a 269% expansion in the outpatient workload in 10 years (1987 to 1997) in Toronto. In our own unit we have seen a 400% growth in the number of clinics for adults with congenital heart disease in the past 10 years. In addition to development of facilities for outpatient surveillance62 adults with congenital heart disease will require inpatient care, reinvestigation, and repeat surgery or catheter intervention.63 ,64
We have used the latest published survival data available. This may not reflect current survival figures but patients who are now 16 years old mostly had operations in infancy 15 years ago and so the choice of references to guide the predictions is justifiable. Improvements in care in the past 15 years mean that even more survivors will turn 16 each year, particularly those with more complex congenital heart diseases that require closer long term follow up and have a higher risk of late complications.
The main limitations of this study are the many assumptions we made as set out above. Our predictions were based on published reports and the methods we used are defined so that others can make any adjustments to the predictions that they deem appropriate. The design of our database means that we have progressive underascertainment in later childhood but we corrected for this and any error would be small. We did not include any cases diagnosed beyond 13 years of age but the effect of this would also be very small. We ignored heart disease other than structural congenital heart disease and we recognise that this leads to an underestimate of total numbers.
It was not the purpose of this study to enter into the discussion about the responsibility for continuing care of adults with congenital heart disease. At present this is undertaken mostly by paediatric cardiologists but in some places there are full time specialists in adult congenital cardiology2 and in others there is a collaboration between paediatric cardiologists and interested adult cardiologists.65 Whatever system is used it is important that appropriate arrangements be made for the handover of care of patients from the paediatric to the adult service.66 ,67
The predicted number of patients graduating to adult follow up each year is relatively small compared with the number of adults with heart disease. However, these patients, their families, and their cardiologists and surgeons have already invested greatly in time, effort, and resources. Rapid recent advances have led to the emergence of adult congenital heart disease as a distinct subspecialty and it is important to be able to predict its future growth to ensure appropriate provision of medical manpower, facilities, and resources for the care of adults with congenital heart disease.
Acknowledgments
We are indebted to Kati Whiteoak and Valerie Sutherland for data collection; to Kathryn Breeze for preparation of the tables, figures, and manuscript; and to the Children's Heart Unit Fund for financial support of the paediatric cardiology database.