Article Text

Abstract
Inflammation and genetics are both prominent mechanisms in the pathogenesis of atherosclerosis and arterial thrombosis. Accordingly, a number of population studies have explored the association of ischaemic heart disease with gene polymorphisms of the inflammatory molecules tumour necrosis factors (TNF) α and β, transforming growth factors (TGF) β1 and 2, interleukin (IL) 1 and its receptor antagonist (IL 1ra), CD14 (the receptor for lipopolysaccharide), P and E selectins, and platelet endothelial cell adhesion molecule (PECAM) 1. Although they are very preliminary and partly conflicting, the data provide some evidence that alterations in the genetics of the inflammatory system may modify the risk of ischaemic heart disease.
- inflammation
- genes
- polymorphisms
- myocardial infarction
- atherosclerosis
- CI, confidence interval
- ECTIM, étude cas-témoin de l'infarctus du myocarde
- IHD, ischaemic heart disease
- IL, interleukin
- IL 1ra, interleukin 1 receptor antagonist
- OR, odds ratio
- PECAM, platelet endothelial cell adhesion molecule
- TGF, transforming growth factor
- TNF, tumour necrosis factor
- VNTR, variable number of tandem repeats
Statistics from Altmetric.com
- CI, confidence interval
- ECTIM, étude cas-témoin de l'infarctus du myocarde
- IHD, ischaemic heart disease
- IL, interleukin
- IL 1ra, interleukin 1 receptor antagonist
- OR, odds ratio
- PECAM, platelet endothelial cell adhesion molecule
- TGF, transforming growth factor
- TNF, tumour necrosis factor
- VNTR, variable number of tandem repeats
Recent attention has focused on the inflammatory component of atherogenesis and acute ischaemia.1,2 Indeed, atherosclerosis is now described as an inflammatory disease1 and flared plaque inflammation is considered a cause of intimal erosion and rupture and therefore of acute ischaemia.2 Studies in vitro and in experimental animals are supported by the clinical finding of increased inflammatory markers in patients with chronic stable angina,3 severe unstable angina, and acute myocardial infarction2 and by the predictive value of such markers for subsequent coronary events.2
Because genetic traits contribute significantly to the global risk of ischaemic heart disease (IHD),2 a number of studies have now addressed the hypothesis that variations in the genetics of the inflammatory system may increase the risk of disease. Differences in the genetic regulation of inflammatory processes might explain why some people but not others develop the disease and why some develop a greater inflammatory response than others.2
Through a Medline search of the literature published from 1992 to July 2001, we reviewed the existing literature linking IHD to gene variants of the inflammatory system, using “inflammation”, “polymorphisms”, “atherosclerosis”, and “myocardial infarction” as keywords. We subsequently focused on cytokines, cell receptors, their antagonists, and adhesion molecules. Bibliographies in the articles provided further references. Relevant journals were also hand searched. With a few exceptions, only case control studies of ≥ 100 cases were included. Whenever possible, studies of atherosclerosis or generic IHD were distinguished from those of acute coronary syndromes (table 1 versus table 24–26), on the assumption that the inflammatory process may differ in the two settings.
Summary of case control studies linking inflammatory gene polymorphisms to myocardial infarction
Summary of case control studies linking inflammatory gene polymorphisms to atherosclerosis
TUMOUR NECROSIS FACTOR α
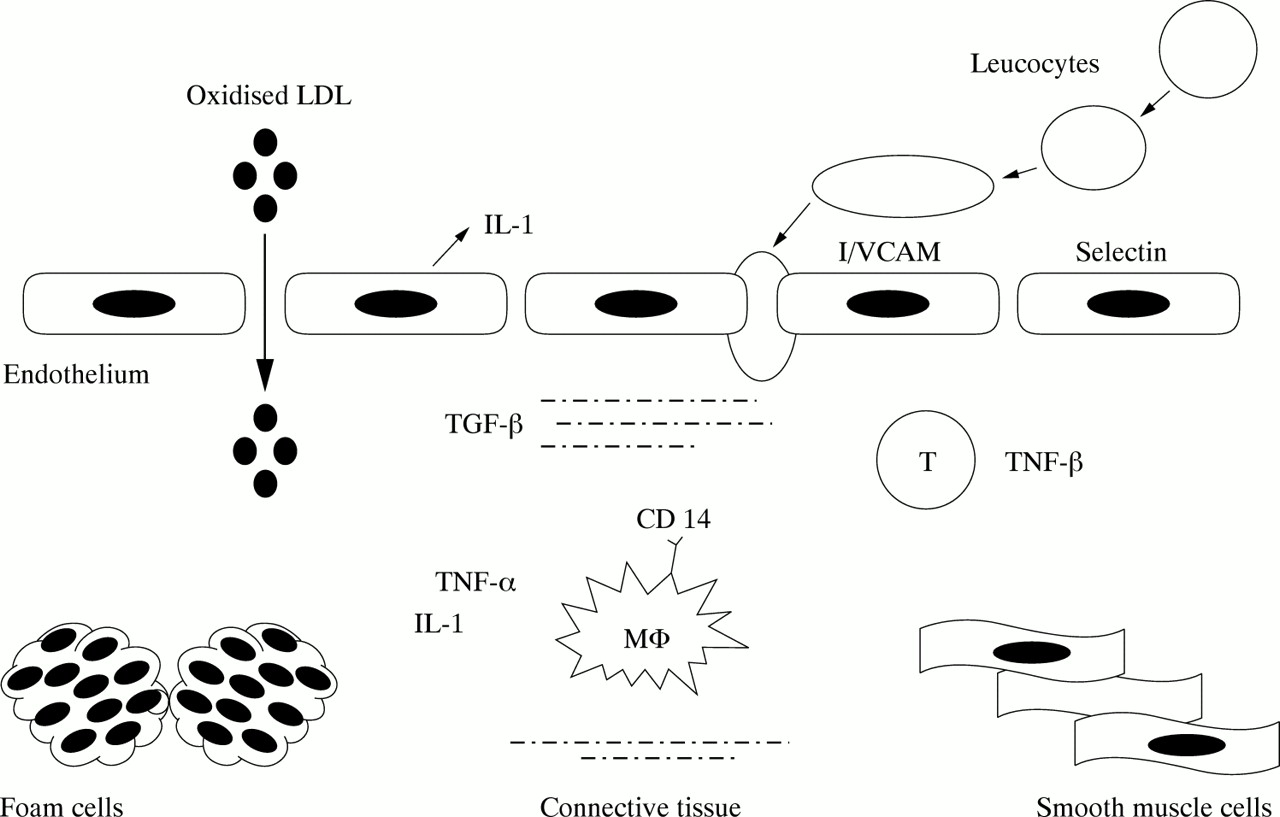
Tumour necrosis factor (TNF) α is a pleiotropic proinflammatory cytokine produced mainly by activated macrophages27 (fig 1). Major effects on the cardiovascular system include increased expression of adhesion molecules and human leucocyte antigen proteins, release of endothelial cytokines and nitric oxide, enhanced vascular permeability, negative inotropism, reduced lipoprotein lipase activity, increased hepatic fatty acid synthesis, involvement in obesity related insulin resistance, and prothrombotic effects (through enhanced expression of plasminogen activator inhibitor 1 and von Willebrand factor and through suppression of the anticoagulant protein C).27 TNF β is synthesised primarily by T cells27 (fig 1).

{kind=link}
Simplified representation of the role of inflammatory molecules in atherogenesis. Injured endothelial cells (for example, by oxidised low density lipoprotein (LDL), cytokines, or lipopolysaccharide) produce cytokines (for example, interleukin (IL) 1), which stimulate the expression of adhesion proteins on the endothelial surface (for example, selectins and cell adhesion molecules). The latter recruit circulating leucocytes into the subendothelial space. Further cytokine release by infiltrated monocyte/macrophages (Mϕ) and T lymphocytes promotes the formation of foam cells and the migration of smooth muscle cells from the media. The proinflammatory effects of IL 1 and tumour necrosis factor (TNF) are counterbalanced by the modulatory effects of transforming growth factor (TGF) produced by connective tissue cells. ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule.
TNF α polymorphisms
The human TNF α gene maps to chromosome 6 (p21.1–21.3) within the human leucocyte antigen complex.27 Data in vitro indicate that a G(–308)A transition modulates transcription.28 Herrmann et al4 screened the coding region of the gene and 1053 base pairs upstream of the transcription starting site: five polymorphisms, including G(–308)A, were identified and assessed in the ECTIM (étude cas-témoin de l'infarctus du myocarde), a study of approximately 650 male survivors of myocardial infarction (mean age 54 years) and 750 control subjects from four regions of France and Northern Ireland. No association was found with either myocardial infarction or coronary artery disease.4 A recent study of 148 survivors of myocardial infarction (< 55 years) and 148 control subjects confirmed the lack of association between –308A and infarction.5 In a study of 674 patients with angiographically confirmed coronary artery stenoses and 1059 controls, the –308A allele again was not associated with the presence or number of stenosed arteries.16 Finally, a recent necropsy series of 700 men also found no significant associations between –308A and either frequency of healed myocardial infarction and coronary thrombosis or number and severity of coronary stenoses.17
TNF β Asp26Thr polymorphism
The TNF β gene is close to and strongly linked with the TNF α gene.27 Braun et al18 investigated a functional polymorphism (Asp26Thr, which correlates with TNF β production) in 199 men with IHD (defined as ≥ 50% diameter stenoses) and 81 control subjects. No association was found with the presence or extent of coronary disease or with previous infarction although, in the patient group, heterozygosity for Thr26 was associated with hyperinsulinaemia.18 Another variant (A252G in intron 2), which correlates with increased TNF β concentrations, was also not associated with myocardial infarction.5 Thus, the TNF polymorphisms explored so far do not appear to be linked to IHD.
TRANSFORMING GROWTH FACTORS β1 AND β2
Transforming growth factor (TGF) is expressed by a wide range of cells, including platelets, endothelial, haematopoietic, and connective tissue cells29 (fig 1). Major functions attributed to TGF β1 are immunosuppression, reduction of inflammation, promotion of wound healing, and regulation of cell proliferation, cell migration, cell differentiation, and extracellular matrix production.29 Various studies suggest an inverse relation between TGF β1 and IHD,29 while others suggest a role in vascular restenosis.30
TGF β1 polymorphisms
The human TGF β1 gene on chromosome 19 (q13.1–13.3)31 was screened in the ECTIM.6 The prevalence of the Arg25Pro (G74C) variant was increased in patients from Belfast (p < 0.01) and Strasbourg (p < 0.05) but not in the two other populations (Lille and Toulouse), although the overall difference was significant (p < 0.05). Among the 374 French patients who underwent coronary angiography, no association was seen between Pro25 and number of ≥ 50% diameter stenoses. Other polymorphisms, including Leu10Pro (T29C), were not associated with disease.6 The latter was evaluated in 315 Japanese survivors of myocardial infarction (234 men) and 591 healthy control subjects.7 On multivariate analysis there was a higher prevalence of the T allele in male patients than in male controls (TT + TC versus CC: odds ratio (OR) 3.5, 95% confidence interval (CI) 2.0 to 6.3, p < 0.0001) but not in women.7 Controls (men and women) with the CC genotype had significantly higher serum TGF β1 concentrations.7
Syrris et al19 also screened the TGF β1 gene and assessed five polymorphisms (including Arg25Pro and Leu10Pro) in 655 patients with IHD (defined as > 30% diameter stenoses, without reference to previous acute events) and 244 patient controls. No significant difference in the distribution of any of these polymorphisms or associated haplotypes was found between patients and control subjects.19 Another common variant in the TGF β1 promoter, a C to T transition at nucleotide –509, assessed in 371 patients also was not associated with the number of ≥ 50% coronary stenoses.20 In summary, the data on TGF β1 are somewhat controversial, with the majority of studies showing no association with IHD; there is, however, a suggestion that certain polymorphisms may increase the risk of infarction within specific ethnic groups.
TGF β2 polymorphism
Biggart et al21 evaluated a TGF β2 gene variant in 101 patients with generic IHD (men < 55 years and women < 60 years of age) and in 100 control subjects with angiographically confirmed normal coronary arteries. The variant was not associated with IHD.
INTERLEUKIN 1 SYSTEM
Interleukin (IL) 1, released by macrophages, platelets, and injured endothelium (fig 1), promotes the interaction of endothelial cells with circulating leucocytes,3,32 induces the activation and proliferation of monocytes/macrophages, and stimulates smooth muscle cell mitogenesis and the synthesis of plasminogen activator inhibitor 1.3,32 It is believed to play a key part in atherogenesis and thrombosis.32 The IL 1 receptor antagonist (IL 1ra) is a soluble antagonist of IL 1 that binds without activity to the IL 1 signalling receptor type I.32 Several autoimmune and inflammatory diseases have been associated with a variable number of tandem repeats (VNTR) of an 86 base pair sequence in intron 2 of the IL 1ra gene.8 There are five alleles for this polymorphism corresponding to two, three, four, five, and six repeats; allele 2 (IL 1ra*2, corresponding to two repeats) has been associated with the severity and extent of diseases in vivo8 and with enhanced production of IL 1β in vitro.33
Polymorphisms of the IL 1 system
The human genes for IL 1α, IL 1β, their receptors, and IL 1ra are clustered on chromosome 2 (q14–q21).8 In an English study,16 allele frequencies of IL 1α (–889), IL 1β (–511 and +3593), and the IL 1ra intron 2 VNTR were measured in 827 healthy blood donors, in 232 patients with angiographically unobstructed coronary arteries (patient controls: 102 from London and 130 from Sheffield), and in 674 patients with single vessel disease (57 and 98, respectively) or multivessel disease (191 and 328, respectively). The IL 1 gene variants were not significantly associated with the presence or extent of disease, whereas homozygosity for IL 1ra*2 was significantly associated with single vessel disease with an OR of 2.8 when data from London and Sheffield were pooled (95% CI 1.4 to 5.7, p = 0.005). Allelic frequencies in patients with multivessel disease were similar to those of healthy and patient controls. To explain the association with single but not with multivessel disease, the authors suggest that the IL 1ra VNTR may act as a “disease modifying” rather than a causative polymorphism.16 In a cohort of 1850 patients another polymorphism in strict linkage disequilibrium with the IL 1ra*2 allele (+2018 in exon 2) was recently associated with protection from coronary restenosis after stenting.22
Two subsequent Italian studies found no significant association between IL 1ra*2 and infarction: the first compared 115 patients with IHD (74 with first myocardial infarction) with 80 matched controls8 and the second compared 148 infarct survivors (men ≤ 45 years and women ≤ 50 years of age) with 153 matched controls.9 In the latter study, the frequencies of IL 1ra*2 were 0.25 in survivors versus 0.25 in controls and those of IL-1ra*1 were 0.72 versus 0.73, respectively. Thus, the association between the IL 1 system and atherosclerosis is probably complex and may vary with clinical phenotype and extent of disease.
CD 14
CD surface molecules mediate cell activation and signalling. In particular, CD14 on blood monocytes mediate monocyte/macrophage activation by lipopolysaccharide.34 Lipopolysaccharide bound to CD14 may contribute to atherogenesis by stimulating macrophages to produce TNF α, IL 1, IL 6, IL 8, IL 12, interferon γ, migration inhibitory factors, chemokines, eicosanoids, and reactive oxygen species, which in turn stimulate the production of a second wave of chemokines, cytokines, and adhesion and signalling molecules35 (fig 1). The mature CD14 protein is present in two isoforms: membrane bound and soluble.34 Endothelial and smooth muscle cells are activated by soluble CD14.36
CD14 polymorphisms
The human CD14 gene maps to chromosome 5.34 Hubaceck et al10 investigated a C to T transition at nucleotide –260 in 178 men who suffered a first myocardial infarction before 65 years of age and 135 controls. The frequency of –260T was 0.49 in patients and 0.35 in controls (OR 1.8, 95% CI 1.3 to 2.5, p = 0.0005); moreover, CD14 receptor density was higher among TT homozygotes than among the other two genotypes (p < 0.003).10 The same dimorphism was investigated by Shimada et al11 in 211 Japanese subjects (128 patients with > 50% stenosis in at least one epicardial artery and 83 healthy controls). In this group, the T allele was strongly associated with myocardial infarction, as TT homozygosity carried an adjusted OR of 3.8 (CI 1.79 to 6.98, p = 0.0001) among the 81 patients with infarction, whereas no association was seen in the 47 patients with stable angina.11
Unckelbach et al12 studied this polymorphism in 2228 men undergoing coronary angiography (mean age 62 years). A healthy control group was not investigated. Overall, no significant association was found between this variant and previous myocardial infarction or severity and extent of coronary disease. However, in the 173 normotensive, non-smoking patients (64 with prior infarction and 109 without) a small increase in the risk of infarction was detected among TT homozygotes (OR 1.6, 95% CI 1.0 to 2.4, p < 0.05).12 The association was stronger in a smaller group of 76 “low risk” patients (30 with previous infarction) older than 62 years (OR 3.8, 95% CI 1.6 to 9.0, p < 0.01).12
Recently, Zee et al,13 in a nested case control study of 387 patients with incident myocardial infarction and an equal number of age and smoking matched control subjects from the Physicians' Health Study, questioned this association because no increased risk was found with the T allele, even in the homozygous state. Overall, these conflicting data may be explained by a number of reasons: the use of subgroup analyses, racial dishomogeneity, and the different definition of phenotype, as infarction at a young age “out of the blue” differs phenotypically from an infarct in an elderly person with multiple risk factors. Further studies should clarify whether the –260T allele of CD14 significantly increases the risk of myocardial infarction among low risk caucasians and the Japanese.
ADHESION PROTEINS
Adhesion of circulating cells to the arterial surface is among the first detectable events in atherogenesis.1 Selectins on the membrane of monocytes, lymphocytes, and endothelial cells enable white cells to roll along the lumen.37 Subsequently, integrins and molecules of the immunoglobulin superfamily (platelet endothelial cell adhesion molecule (PECAM) 1, intercellular adhesion molecule 1, and vascular cell adhesion molecule 1) on leucocytes and endothelial cells allow white cells to attach firmly to the vessel wall, enabling them to cross it1 (fig 1). P selectin (CD62P) is stored in endothelial Weibel Palade bodies and platelet α granules. It appears on the endothelial and platelet surface within minutes after stimulation by IL 1.37 Its expression is increased in atherosclerotic plaques38 and high plasma concentrations of soluble P selectin are found in patients with unstable angina.39 E selectin (CD62E) is transiently expressed on endothelial cells within hours after stimulation by TNF α, IL 1, and lipopolysaccharide.40 L selectin is constitutively expressed by all leucocytes while PECAM 1 (CD31) is constitutively expressed by endothelial cells, bone marrow stem cells, platelets, and most circulating leucocytes.41 Soluble PECAM 1 is increased in patients with acute myocardial infarction.42
P selectin Pro715 polymorphism
Human genes of the selectin family are clustered on the long arm of chromosome 1 (q21–q24).43 Thirteen P selectin gene polymorphisms were assessed in the ECTIM.14 The Pro715 allele of the missense Thr715Pro variant was globally less frequent in patients than in control subjects (p < 0.002 and p < 0.02 after correction for number of tests), with an OR for infarction of 0.7 (95% CI 0.5 to 0.9).14 The lower Pro715 frequency among infarct survivors was later confirmed in the ECTIM extension of approximately 600 patients (29% women) and 600 control subjects from Belfast and Glasgow (p = 0.054).44 The Pro715 allele may thus confer protection against myocardial infarction. Functional data on this polymorphism are still unclear.14
E selectin polymorphisms
The E selectin A561C transversion (Ser128Arg) was investigated by Wenzel et al in two studies.23,24 The variant is thought to modify the molecule's interaction with its ligands.45 A first study found a higher Arg128 prevalence among 97 patients ≤ 50 years old with severe coronary or peripheral atherosclerosis than among control subjects (allele frequency 0.16 v 0.09, p = 0.02) with a stronger association among the 40 patients who were ≤ 40 years old (Arg128 frequency 0.21, p = 0.003).23 In a second study, a number of polymorphisms of E, P, and L selectin and of intercellular adhesion molecule 1 and vascular cell adhesion molecule 1 were assessed in 99 patients (defined as above) and 100 age matched healthy volunteers.24 Three E selectin polymorphisms (Arg128, 98T, and Phe554) were all significantly associated with early onset atherosclerosis (but without correction for repeated testing). In patients and control subjects, the Arg128 and 98T alleles were correlated (r = 0.9, p < 0.001) while the frequency of Phe554 was 0.06 v 0.02 (p = 0.02).24 A higher frequency of Arg128 was also observed by Ye et al25 among 82 patients with > 50% diameter-stenoses at coronary angiography than among 71 control subjects with normal angiograms (0.20 v 0.11, p < 0.05). In contrast, in ECTIM, Arg128 was not significantly associated with myocardial infarction.14 Thus, the E selectin Arg128, 98T, and Phe554 alleles may increase the risk of atherosclerosis (but not necessarily of infarction), especially in younger patients.
PECAM 1 polymorphisms
The human PECAM 1 gene maps to 17q23.46 Two polymorphisms, Leu125Val (leading to an immunological difference in the mature protein41) and Ser563Asn, were evaluated in 98 patients (25–50 years old) with ≥ 50% coronary artery stenoses and 103 healthy control subjects.26 Each variant was more frequent in patients (Val125 0.65 v 0.51, Asn563 0.63 v 0.50, p < 0.05) as was the homozygous combination Val125/Asn563 (43% v 26%, p < 0.05).26 Val125 was also investigated by Gardemann et al15 in 2500 German men undergoing diagnostic angiography, grouped according to previous myocardial infarction and the presence and severity of coronary disease. A healthy control group was not included. No evidence of association was found when patients without infarction or significant disease (n = 488) were compared with the 1170 patients with previous infarction. Among those with coronary atherosclerosis, however, Val125 was more frequent in the non-diabetic and normotensive group (n = 809, OR = 1.54, p = 0.035).15 Thus, as for E selectin, the PECAM 1 Val125 and Asn563 alleles may increase the risk of atherosclerosis (but not necessarily of infarction), especially in patients with a low atherosclerotic risk profile.
DISCUSSION
Interpretation of population association studies: susceptibility, linkage or stratification?
The reviewed studies, summarised in tables 1 and 2, are all conceived to show a relation between allele “A” and a multifactorial disease “D” whenever the frequency of A differs between patients and control subjects.47 An association between A and D, however, may arise for at least three reasons: firstly, A truly affects the susceptibility to D; secondly, A is closely “linked” to the real allele involved in the pathogenesis of D, producing an association known as “linkage disequilibrium”; and thirdly, people with D or without D are genetically different and coincidentally also differ in the frequency of A (population stratification).47 In the specific case of myocardial infarction, given its high early mortality, possible alleles associated with rapidly fatal infarction may be underestimated among survivors (survival bias). Since the precise role of A in the pathogenesis of D cannot be fully shown by population association studies, these should be considered at best as hypothesis generating.48
Small biological effect of single polymorphisms: importance of clinical phenotype
Several of the reviewed polymorphisms (for example, those for TNF α and β, TGF β1, IL 1ra, CD14, and E selectin) have been found to be functional—that is, to have direct effects on gene transcription and protein function. However, although allele A may clearly be associated with protein function or concentrations, and protein concentrations with disease, it still may not be possible to relate a given genetic variant to disease, as an individual polymorphism contributes only a fraction to the entire heritable variance in protein concentrations.49 Additionally, the small contribution of a single novel polymorphism to the overall risk of a multifactorial disorder such as IHD may be obscured by the presence of one or more dominant classical risk factors.49 Several examples from this review support this possibility (for example, the association between CD14 variants and myocardial infarction in normotensive and non-smoking patients or in the generally low risk Japanese population, or between E selectin or PECAM 1 polymorphisms and coronary atherosclerosis in young, non-diabetic, normotensive groups).
Prospectively designed studies and the analysis of haplotypes may overcome some of the limitations of population association studies.50 Another way to confront these limits is through careful phenotypic characterisation of patients, since a new genetic risk factor is more likely to emerge within homogeneous groups of patients in whom it has a similar role. Identifying such homogeneous groups may be difficult and may require rigorous control not only of age, sex, race, and ethnic grouping but also of clinical features and biochemical markers linked to specific pathogenetic mechanisms.
Clinical implications and conclusions
Given the hurdles that riddle the molecular genetics of multifactorial disorders such as myocardial infarction and atherosclerosis, it is surprising in a way that a number of the reviewed studies have yielded “positive results”. It is well appreciated, however, that it is much easier to publish a small study with a significant result rather than a large one where no effect on risk is observed, a problem known as “publication bias”.
For myocardial infarction, the data suggest protection in carriers of the P selectin Pro715 allele and increased risk within specific groups carrying TGF β1 and CD14 variants. For atherosclerosis, the IL 1ra intron 2 VNTR alleles may modulate the extent of disease, whereas certain polymorphisms of E selectin and of PECAM 1 appear to increase the risk, especially in otherwise low risk groups. The association of TGF β1 Pro25 with myocardial infarction and, conversely, of E selectin Arg128, PECAM 1 Val125, and IL 1ra*2 with atherosclerosis raises the possibility that the genetic effects on infarction (possibly through haemostasis or plaque fissure) may be distinct from those on atherosclerosis.
With the advent of DNA microchips, which allow a simultaneous and rapid assessment of multiple genetic variants,51,52 and with further studies on the functional relevance of the single polymorphism, it should be possible to draw a more accurate profile of the inflammatory gene variants involved in IHD.
REFERENCES
Linked Articles
- Miscellanea
- Miscellanea