Article Text

Abstract
Background: Telomeres are shorter in subjects with coronary artery disease (CAD) and may indicate premature biological ageing. However, whether shorter telomeres are a primary abnormality or secondary to the disease is unclear.
Objective: To investigate whether shorter telomeres are a primary abnormality or secondary to CAD, telomere lengths in healthy young adults with contrasting familial risk of CAD were compared.
Design: Case–control study.
Methods: Mean telomere restriction fragment (TRF) length in DNA from circulating leucocytes was determined by Southern blotting in 45 healthy offspring of subjects with premature CAD (case offspring) and 59 offspring from families without such a history (control offspring). Correlation in mean TRF length was also assessed in 67 offspring–parent pairs.
Results: On average, a decrease of 27.5 (10.7) bp in mean TRF per year of age was found. The unadjusted mean TRF length was 6.34 kb (95% CI 6.13 to 6.55) for case offspring and 6.75 kb (95% CI 6.57 to 6.94) for offspring of controls (p = 0.004). The adjusted difference in mean TRF between case and control offspring was 472 bp (95% CI 253 to 691, p<0.001), equivalent to about 17 years of age-related attrition in telomere length. Furthermore, there was a significant positive correlation in mean TRF length between offspring and their parents (r = 0.37, p = 0.002).
Conclusion: These findings suggest that inheritance of shorter telomeres is associated with increased familial risk of CAD. They support the hypothesis that telomere length is a primary abnormality involved in the pathogenesis of CAD.
Statistics from Altmetric.com
Telomeres are the extreme ends of eukaryotic chromosomes and comprise a highly repetitive tandem repeat sequence (in humans, TTAGGG). They function in part to maintain genomic integrity and are also involved in cell cycle regulation.1 2 Mean telomere length varies considerably between individuals at birth.3 Furthermore, because DNA polymerase cannot extend to the ends of chromosomes, in the absence of telomerase some telomeric DNA is lost during each mitotic division, a phenomenon referred to as the “end-replication problem”. Olovnikov was the first to suggest that this is a potential mechanism for a “biological clock” capable of limiting cellular lifespan,4 a concept that has since been supported experimentally.5 6 Thus telomere length is now regarded as a marker of cellular ageing, with shorter telomeres indicating increased cellular age and reduced replicative capacity.7
We have previously shown that subjects with coronary artery disease (CAD) or premature myocardial infarction (MI) have shorter mean leucocyte telomeres than normal subjects matched for age and gender.8 9 Furthermore, shorter mean leucocyte telomere length precedes the clinical development of ischaemic heart disease.10 Cellular senescence is an important feature of the atherosclerotic plaque.11–13 In addition, telomere disruption leads to changes in expression levels of genes implicated in atherosclerosis.14 This has led to the hypothesis (the “telomere hypothesis”) that shorter telomeres may be causally implicated in the pathogenesis of CAD.10
Both CAD and MI have a substantial but hitherto poorly characterised genetic basis.15 The increased risk of CAD in an offspring of a subject with known CAD ranges from two- to sevenfold depending on the age of onset in the parental generation.16 Mean telomere length also exhibits strong genetic determination.17–19 If the telomere hypothesis is correct, then one would expect the mean telomere length, on average, to be shorter in the offspring of subjects with a history of CAD than in subjects without such a history, and that this would be the case before the onset of disease in the offspring. In this study we therefore compared mean leucocyte telomere length in healthy young adult offspring of people with and without a history of CAD.
SUBJECTS AND METHODS
Subjects
Study subjects were recruited in two phases. In the first phase (phase 1) we studied 46 young healthy men: 22 with a strong family history of MI (validated MI <50 years of age in a parent) (case offspring) and 24 controls with no family history of CAD under the age of 65 in the preceding two generations (control offspring). The second phase (phase 2) comprised 58 young healthy adult subjects: 23 (case offspring) with a parent with severe premature CAD confirmed on coronary angiography (>50% stenosis in all three vessels before the age of 65 years) and 35 (control offspring) with no history of CAD in either parent. Case offspring were all identified and recruited through their affected parent who was treated in our hospital. Control offspring were recruited in phase 1 mainly from healthy staff volunteers and in phase 2 the majority were recruited from a continuing family-based study of subjects representative of the general population.20 Where possible, DNA was also collected from the parental generation (affected parent in the case offspring and one parent in the control offspring).
Demographic information including cardiovascular risk factor status was recorded for all subjects. History of hypertension, hypercholesterolaemia and smoking was based on patient self-reporting. Exclusion criteria for recruitment of both case offspring and control offspring were diabetes mellitus, malignancy, concomitant inflammatory conditions (including asthma) and renal impairment (serum creatinine >150 µmol/l). None of the offspring came from families known to have familial hyperlipidaemia. All subjects were Caucasian of northern European origin and the study was approved by the Leicestershire Research Ethics Committee.
Measurement of mean leucocyte telomere length
Mean leucocyte telomere length was measured as previously described8 10 18 and blinded to the clinical data. Briefly, leucocyte DNA was extracted from peripheral blood samples, digested in 2 μg aliquots overnight at 37°C with 15U RsaI and HinfI (Invitrogen, Paisley, UK), and resolved by electrophoresis on a 0.5% agarose gel (850 V/H). DNA was transferred to Hybond-N+ membrane (GE Healthcare, Amersham, Bucks, UK) before being hybridised at 42°C overnight to a 3′-end labelled 32P-(AATCCC)3 oligonucleotide telomere probe. After washes in 3× standard saline citrate/0.1% sodium dodecyl sulphate at room temperature, telomere smears were digitised using a Molecular Dynamics Typhoon Phosphorimager. ImageQuant version 5.1 software was used to analyse telomeric smears, and the mean size of the telomere restriction fragment (TRF) was estimated using the formula TRF = (ODi)/(ODi/MWti), where ODi is the optical density at a given position on the gel, and MWti is the molecular weight at that position. A calibrator sample was included on every gel to allow correction for interassay variability.
Statistical analysis
Demographics are presented for cases and controls. A χ2 test of association (categorical data) or t-test (continuous data) was used to compare the two groups.
A generalised linear model in SAS version 9.1 (SAS Institute, Cary, SC, USA) was used to compare the mean telomere length between the offspring of cases and controls, adjusting for age, sex, body mass index (BMI) and phase of recruitment. Linear regression was used to investigate the independent relationship between telomere length and smoking and BMI. For analysis purposes, smoking status was categorised into either “non-smoker” or “current/ex-smoker”. All continuous variables were normally distributed.
RESULTS
Demographics
Offspring of cases and controls were reasonably well matched for age and sex (table 1). There were 67 non-smokers, 5 ex-smokers and 32 current smokers in the cohort. There were slightly more current/ex smokers in the offspring of cases, although this did not reach significance. Mean BMI was significantly higher in the offspring of cases than in the offspring of controls. Four women (one case offspring, three control offspring) were taking the oral contraceptive pill and one subject was taking Seroxat. None of the subjects were taking statins or any other cardiovascular drugs.
Telomere length in healthy offspring of cases and controls
There was a significant age-related decrease in mean TRF length in case offspring, but not control offspring (mean (SE): cases 33.3 (14.3) bp/year, p = 0.025; controls 14.9 (15.2) bp/year, p = 0.33). The difference between these (the slopes) was non-significant (p = 0.382). The average decrease was 27.5 (10.7) bp (p = 0.012)
The unadjusted mean TRF length was 6.34 kb (95% CI 6.13 to 6.55 kb) for case offspring and 6.75 kb (95% CI 6.57 to 6.94 kb) for control offspring of controls (p = 0.004) (fig 1). After adjustment for age, sex, BMI and recruitment phase, the difference in mean TRF length between offspring of cases and controls was 472 bp (95% CI 253 to 691 bp), p<0.001).
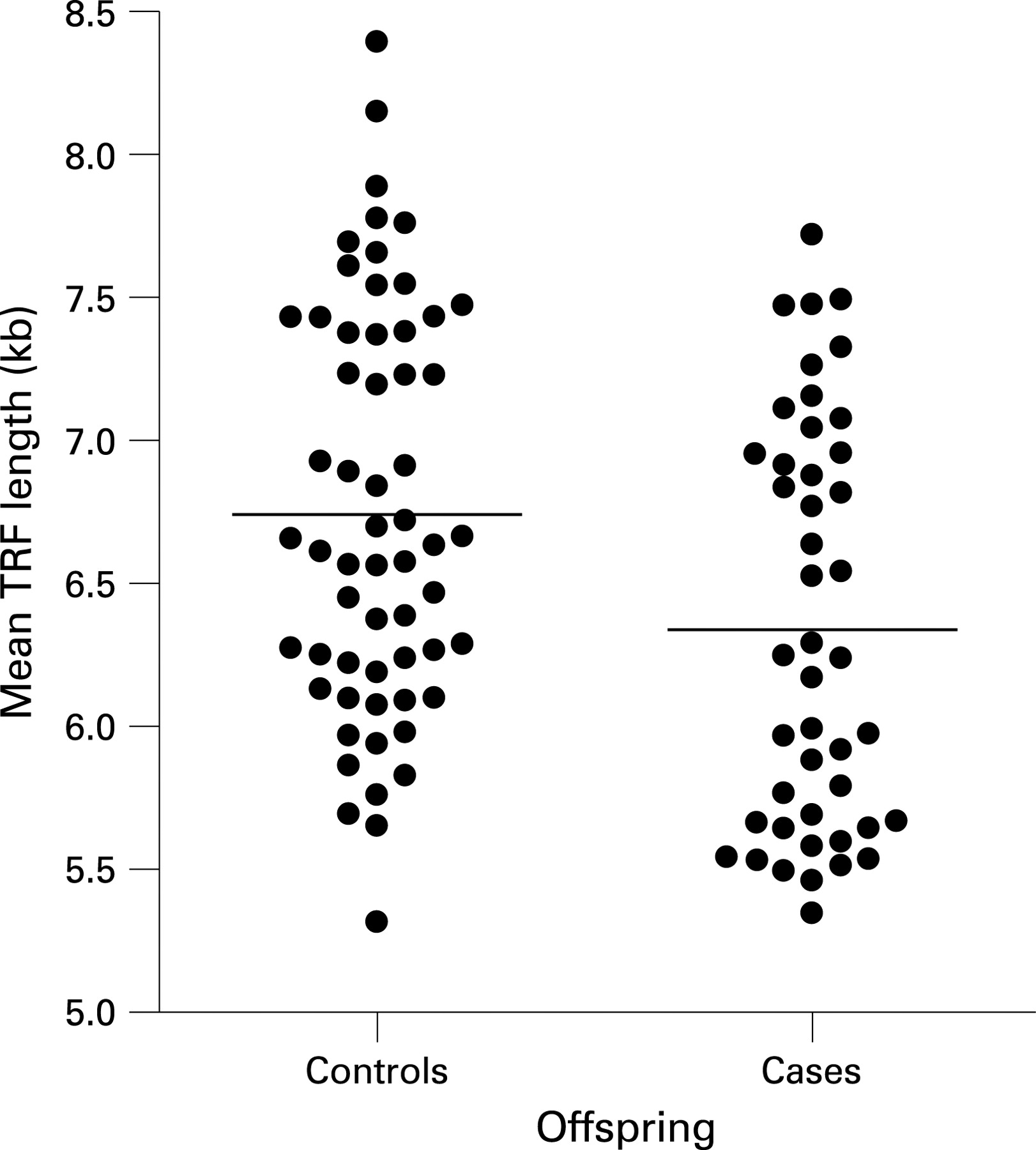
There was no independent relationship of either BMI (p = 0.921) or smoking status (p = 0.947) with telomere length. Likewise, among the current smokers there was no relationship between telomere length and pack-years (p = 0.940) accounting for other variables. Total and high-density lipoprotein (HDL) cholesterol values were available for offspring from phase 1. There was no association between TRF length and either total (p = 0.467) or HDL cholesterol (p = 0.175), accounting for age and case–control status.
Correlation of telomere length between parents and offspring
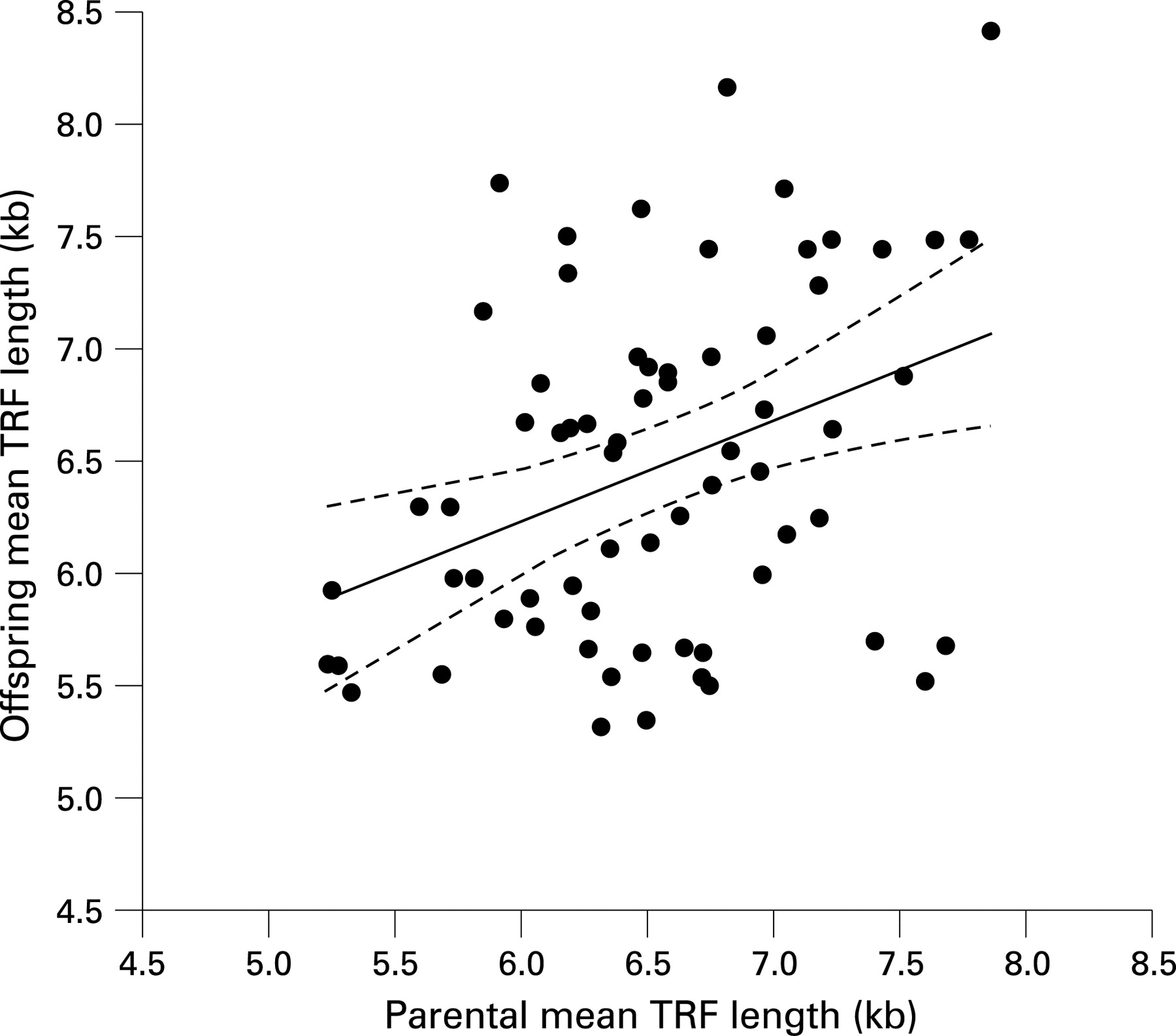
Telomere lengths were available for a parent of 67 of the offspring. There was a significant positive correlation in mean TRF length between offspring and parents (r = 0.37, p = 0.002) (fig 2).

{kind=link}
{kind=link}
DISCUSSION
In this study we show, for the first time, that healthy young adult subjects with contrasting familial risk of CAD differ in their mean telomere lengths. Those with a strong family history of CAD had significantly shorter leucocyte telomeres than those without such a history. The findings support the association of shorter telomeres with CAD and suggest that they may be causally involved.
Several cross-sectional studies have shown an association between shorter telomeres and CAD8 9 21; however, it is not possible to dissect cause and effect from such studies. Recently, prospective studies have provided additional support for an association between telomere length and subsequent development of clinical coronary disease and mortality from coronary heart disease.10 22 However, since these latter studies analysed telomeres in middle-aged subjects it is difficult even here to wholly exclude the possibility of shorter telomeres being secondary to existing occult CAD. Here, by using family history to partition those with and without risk and examining healthy subjects at a much younger age, it is unlikely that the finding of shorter telomeres in those with increased risk is a consequence of significant existing but undetected disease. The findings indicate a primary association between shorter telomeres and CAD.
As for any putative CAD risk factor, the partitioning of telomere length between offspring of cases and controls is not (and not expected to be) absolute. However, the observed average difference in mean telomere length between offspring of subjects with and without CAD of 472 bp is substantial in biological terms. The average rate of telomere attrition for both groups combined was 27 bp/year. Thus, the mean telomere length in the offspring of cases is similar to that in controls approximately 17 years older. This compares with differences in telomere length, equating to 9–12 years in previous case–control studies of subjects with and without CAD or premature MI.8 9 If shorter telomeres are indeed causally associated with CAD, then the larger difference seen here may reflect the more extreme contrast between the groups based on their familial risk. However, this estimation should be interpreted with some care as our estimate of the average age-related decline in telomere length in the present study is based on a rather small sample size and a relatively small age range. Indeed, in control offspring, although there was a trend towards a decline in telomere length with age it did not reach statistical significance probably owing to sample size. Other studies have found age-related attrition rates of anywhere between 15 and 35 bp/year.8–10 18 19 21 Irrespective of the precise estimate for the difference in case and control offspring telomere length in terms of equivalence to age-related attrition, the findings do unequivocally show that mean telomere lengths of the offspring of cases are substantially shorter than those of the offspring of control subjects.
Our study does not identify the reason behind the difference in mean telomere length in the offspring of subjects with and without CAD. Specifically, given the difference, albeit non-significant, in age-related attrition rates of telomere length between case and control offspring, we cannot exclude the possibility of a role for postnatal factors in explaining at least some of the difference. Our study had limited power to exclude an effect of factors such as smoking or dyslipdaemia. However, in much larger studies9 10 we have not observed an association between telomere length and either lipid traits or smoking and even if these factors do affect telomere length, it is unlikely that they can account for the differences seen between the offspring groups. On the other hand, mean telomere length has a strong genetic determination17–19 and it is likely that at least a proportion of the difference has a genetic basis. Indeed, and consistent with previous studies, we found a significant correlation in telomere length between parents and offspring. Our findings therefore raise the intriguing possibility that, in addition to specific genes, a part of the genetic contribution to CAD is from a more global structural property of the genetic material—namely, telomere length. In this context, it is relevant to note that there is good correlation of telomere lengths in different tissues,23 24 and a particularly strong correlation between mean leucocyte telomere length and telomere length in vascular tissues has recently been shown.13
Cellular senescence is a characteristic feature of atherosclerotic plaque and has an important role in its pathogenesis.11–13 Telomere shortening is an important contributor to the development of the senescent phenotype in vascular endothelial and smooth muscle cells.13 14 21 Therefore people who are born with, or acquire, shorter telomeres may reach this state earlier. This might explain the association between shorter telomeres and increased risk of atherosclerosis.
Our findings suggest that a better understanding of the factors that influence telomere length and dynamics may have fundamental and important implications for tackling atherosclerosis and CAD. If telomere shortening accentuates atherosclerosis by triggering senescence then mechanisms that counteract this process may be beneficial. This model could help explain the observation that treatment with statins preferentially attenuates the risk of CAD in subjects with shorter telomeres.10 Statins upregulate the expression of the telomere-associated protein, TRF2, which may stabilise telomere structure.25 Other treatments that affect telomere attrition may have similar beneficial effects.
In summary, mean telomere length is shorter in healthy adult offspring of people with premature CAD. The finding provides strong supportive evidence for the telomere hypothesis and suggests that shorter telomere length is causally associated with increased susceptibility to CAD.
The study was approved by the Leicestershire Research Ethics Committee.
Acknowledgments
NJS holds a personal chair from the British Heart Foundation. We thank subjects who participated in the study and colleagues who helped in recruitment of the subjects.
REFERENCES
Footnotes
Funding: Supported by grants from the British Heart Foundation and the Sir Jules Thorn Trust.
Competing interests: None declared.