Article Text

Abstract
End stage renal disease is associated with a very high risk of premature cardiovascular death and morbidity. Early stage chronic kidney disease (CKD) is also associated with an increased frequency of cardiovascular events and is a common but poorly recognised and undertreated risk factor. Cardiovascular disease in CKD can be attributed to two distinct but overlapping pathological processes, namely atherosclerosis and arteriosclerosis. While the risk of athero-thrombotic events such as myocardial infarction is elevated, arteriosclerosis is the predominant pathophysiological process involving fibrosis and thickening of the medial arterial layer. This results in increased arterial stiffness causing left ventricular hypertrophy and fibrosis and the exposure of vulnerable vascular beds such as the brain and kidney to high pressure fluctuations causing small vessel disease. These pathophysiological features are manifest by a high risk of lethal arrhythmia, congestive heart failure, myocardial infarction and stroke. Recent work has highlighted the importance of aldosterone and disordered bone mineral metabolism.
- Endothelium
- Renal Disease
Statistics from Altmetric.com
Introduction
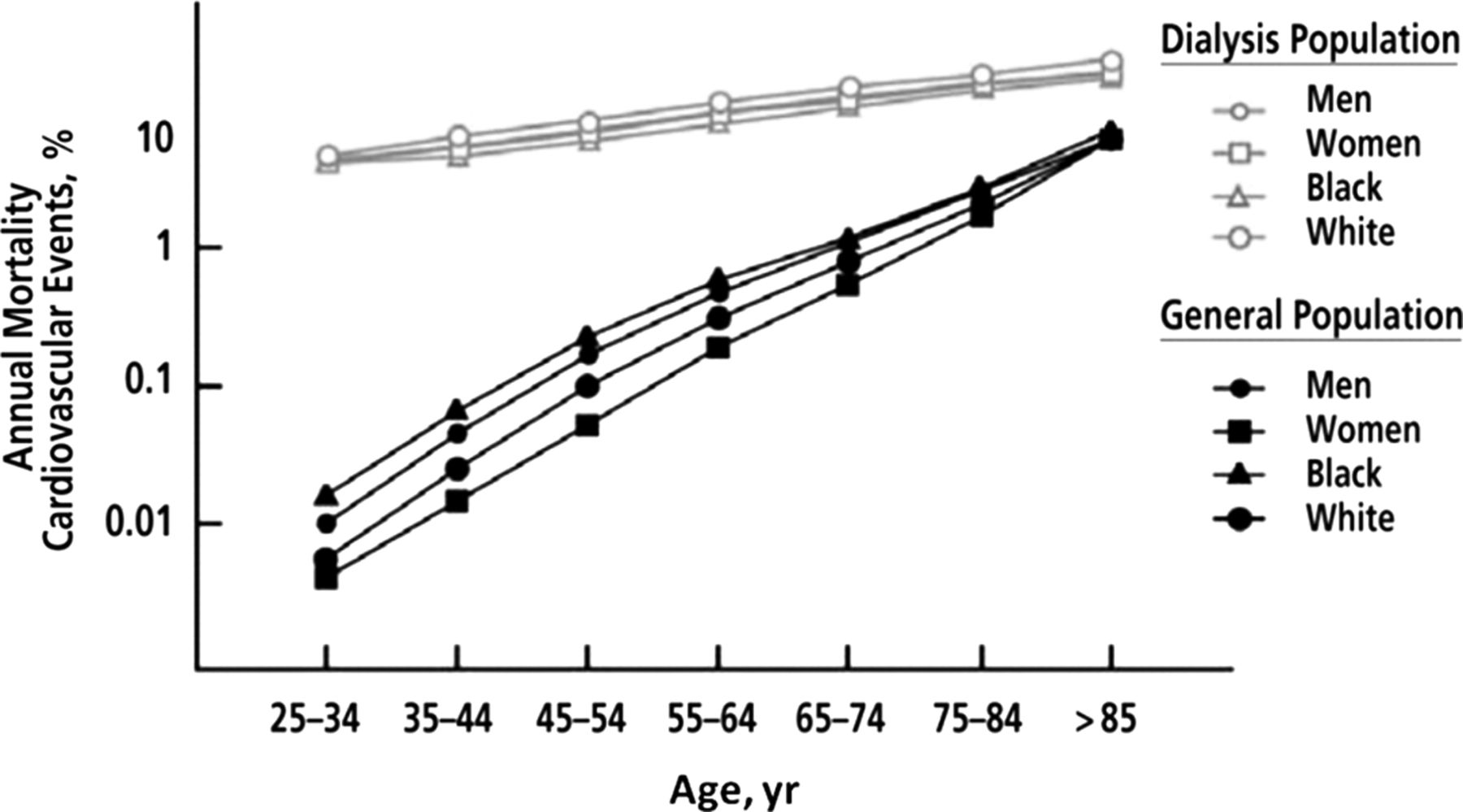
Following the introduction of dialysis as treatment for patients with end stage renal disease (ESRD), it became evident that cardiovascular disease is a major cause of premature mortality.1 Compared with the general population, the cardiovascular death rate is at least 10 times higher and in young subjects this risk is more than a 100-fold (figure 1).2 It is now appreciated that early stage chronic kidney disease (CKD) is also a risk factor for cardiovascular disease. The severity of renal disease is classified into five stages on the basis of estimated glomerular filtration rate (eGFR) and markers of kidney damage (table 1).3 Use of the Modification of Diet in Renal Disease and, more recently, the CKD Epidemiology Collaboration formulae to accurately determine eGFR has facilitated research demonstrating a graded association of milder forms of CKD with cardiovascular risk. With a high and rising prevalence (13% in the USA in 2007), early stage CKD is a major public health problem due largely to the increased risk of cardiovascular disease; patients with early stage CKD are far more likely to die from cardiovascular causes than to progress to ESRD.4 ,5
Stages of CKD as defined by the NKF K/DOQI criteria3
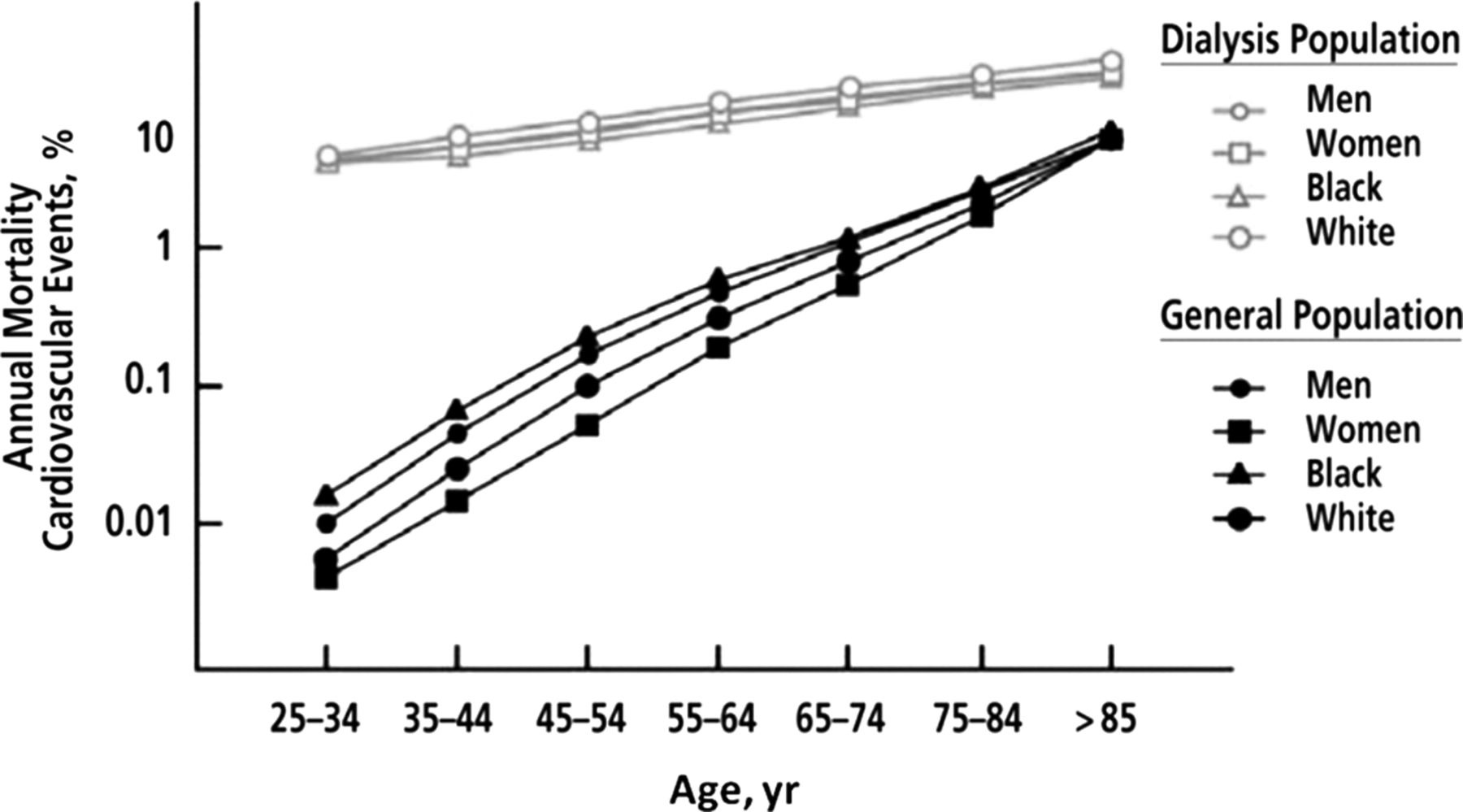
Annual cardiovascular mortality (death as a result of arrhythmias, atherosclerotic heart disease, cardiac arrest, cardiomyopathy, myocardial infarction or pulmonary oedema) in the US general population compared with patients with end stage renal disease who were treated with dialysis by age, sex and racial group. Adapted from Foley et al2 with permission from the National Kidney Foundation.
Several large studies including a meta-analysis of general population cohorts of over 4 million subject years have shown an independent, graded, inverse correlation between eGFR and cardiovascular event rates.6–8 The ‘threshold’ eGFR at which cardiovascular risk first rises is unclear. While expert consensus proposes a level of <60 ml/min/1.73 m2,3 ,9 two recent studies with long-term follow-up (median follow-up 7.9 and 10 years respectively) suggest that the increased cardiovascular mortality may begin even earlier, perhaps at eGFR levels <90 ml/min/1.73 m2.8 ,10 In a recent meta-analysis of the relationship between eGFR and cardiovascular risk, it was estimated that a 30% lower eGFR was consistently associated with a 20%–30% higher risk of major vascular events and all-cause mortality.11 If causal, this would imply that up to 10% of vascular events in middle age and 20% in old age might be attributable to reduced eGFR.
There are numerous data showing that proteinuria also has prognostic implications as a cardiovascular risk factor.12 A powerful, independent and graded association exists between the degree of renal albumin loss and cardiovascular risk in many populations including hypertensives, diabetics, those with established vascular disease and the general population.13 ,14 Cardiovascular risk begins to increase even within currently defined normal levels of albuminuria and below those that can be detected on standard urinary dipstick testing.14 In a CKD Prognosis Consortium meta-analysis of data from general population studies, urinary albumin to creatine ratio was independently associated with adjusted risk of all-cause and cardiovascular mortality with a log-linear relationship without any evidence of a threshold effect.8 Albuminuria, together with eGFR, exerted a multiplicative effect on the risks of all-cause and cardiovascular mortality.
The increasing emphasis placed on the early detection of CKD worldwide provides a potential opportunity to reduce cardiovascular risk. The development of risk lowering therapies is, however, hindered by the fact that although the epidemiological association between CKD and cardiovascular disease is robust, the pathophysiological mechanisms underlying this relationship are not well understood. There are a number of lines of evidence suggesting that modification of conventional atherosclerotic risk factors may not be adequate in CKD. First, although in CKD there is a clustering of classical risk factors such as hypertension and diabetes mellitus, they perform very poorly in the prediction of cardiovascular risk in this population, particularly in late stage CKD.15 Indeed, in ESRD there is an inverse relationship between cardiovascular event rates and conventional risk factors such as total cholesterol, obesity and even blood pressure (BP). These paradoxical associations may, at least in part, be explained by ‘reverse causality’, a feature of chronic disease malnutrition–inflammation syndromes.16 Second, epidemiological studies suggest that while approximately 40% of all deaths in patients with ESRD are due to cardiovascular disease, the majority of cardiovascular deaths are attributable to sudden cardiac death, arrhythmia or congestive heart failure (figure 2) with relatively few from vasculo-occlusive events such as myocardial infarction.17 Heart failure is a major cause of morbidity and mortality in both early stage CKD and ESRD with incident rates three to four times higher than in non-CKD subjects.18 Thus, it appears that it is not atheromatous coronary artery disease but myocardial disease (left ventricular (LV) hypertrophy with fibrosis and diastolic dysfunction, often accompanied by systolic dysfunction) which is the principal cause of cardiovascular death and disease in CKD. LV hypertrophy is present from the earliest stages of progressive renal disease and has a prevalence of over 70% on cardiac MRI in patients with ESRD.19 ,20 Epidemiological data from the US Renal Data System 2011 report are consistent with this paradigm (figure 2)17 as is the finding in the SHARP (Study of Heart And Renal Protection) trial of lipid lowering therapy in CKD in which low-density lipoprotein reduction resulted in a significant decrease in major atherosclerotic events, but no significant reduction in total vascular mortality.21 These data are similar to those seen in lipid lowering trials in heart failure;22 in both conditions, most deaths appear to be caused by pump failure or arrhythmia rather than coronary events.17

Causes of death in prevalent US dialysis patients. AMI; acute myocardial infarction; CHF, congestive heart failure; CVA, cerebrovascular accident. Figure reproduced from Chapter 4 of the US Renal Data System annual report (2011).17
Atheroma in CKD
Examination of the arterial system of patients with CKD reveals two distinct but overlapping arterial pathologies, atherosclerosis and arteriosclerosis.23 While atherosclerosis is primarily an intimal disease, patchy in distribution and occurring preferentially in medium-sized conduit arteries, arteriosclerosis is a diffuse disease of the media which leads to increased arterial stiffness (see below). The morphological characteristics of atheroma in patients with CKD are distinct and include increased plaque calcification and increased intimal and medial thickness.24 The prevalence of atheromatous disease in CKD is high, although it is not clear whether this is a direct result of renal dysfunction or the clustering of risk factors such as hypertension, diabetes and inflammation which invariably accompany CKD. A study of carotid intima-media thickness from Finland showed a fourfold greater plaque score in CKD patients (predialysis, dialysis and post-transplant) compared with controls.25 In a recent Canadian study, compared with a reference control group the adjusted relative risk of myocardial infarction was 1.4 in patients with non-diabetic CKD and 2.7 in diabetic stages 3 and 4 CKD compared with 2.0 for diabetes and 3.8 for subjects with previous myocardial infarction.26 When severe proteinuria was present, however, the relative risk of myocardial infarction in non-diabetic CKD equalled that of the diabetic population. There is also an increased prevalence of peripheral and cerebral vascular disease in CKD according to data from the US Renal Data System (figure 3) though it must be remembered that these data are not controlled for comorbid states known to be strongly associated with atheroma such as diabetes and hypertension.17 There is clear evidence of an adverse interaction between CKD and major atherosclerotic events; outcomes after acute coronary syndrome and stroke are much worse in CKD than in the general population.27–30 In ST elevation myocardial infarction, for example, patients on dialysis (treated in the preprimary angioplasty era) had a 1 year death rate of almost 60% with a 1 year cardiac mortality of 41%.31
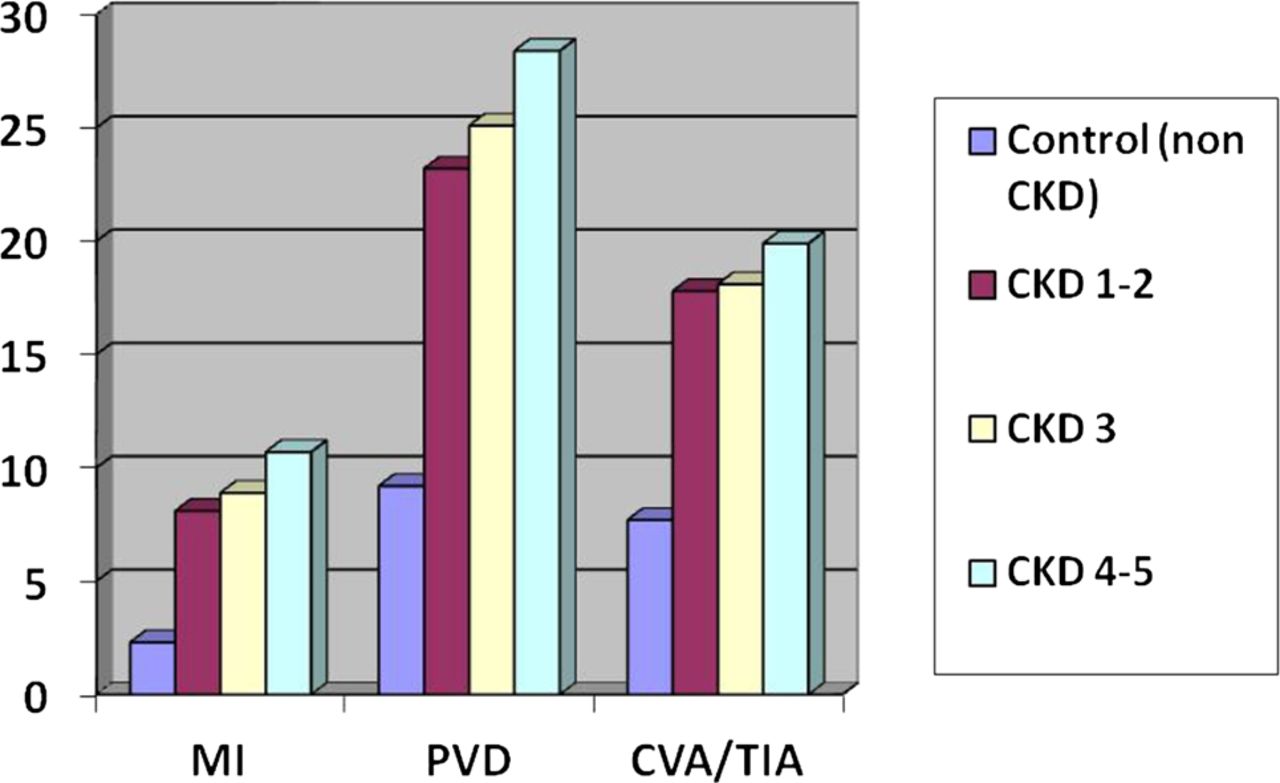
Cardiovascular disease prevalence rates (%) by CKD stage for common cardiovascular diseases. CKD, chronic kidney disease; CVA, cerebrovascular accident; MI, myocardial infarction; PVD, peripheral vascular disease; TIA, transient ischaemic attack. Figure reproduced from Chapter 4 of the US Renal Data System report (2011).17
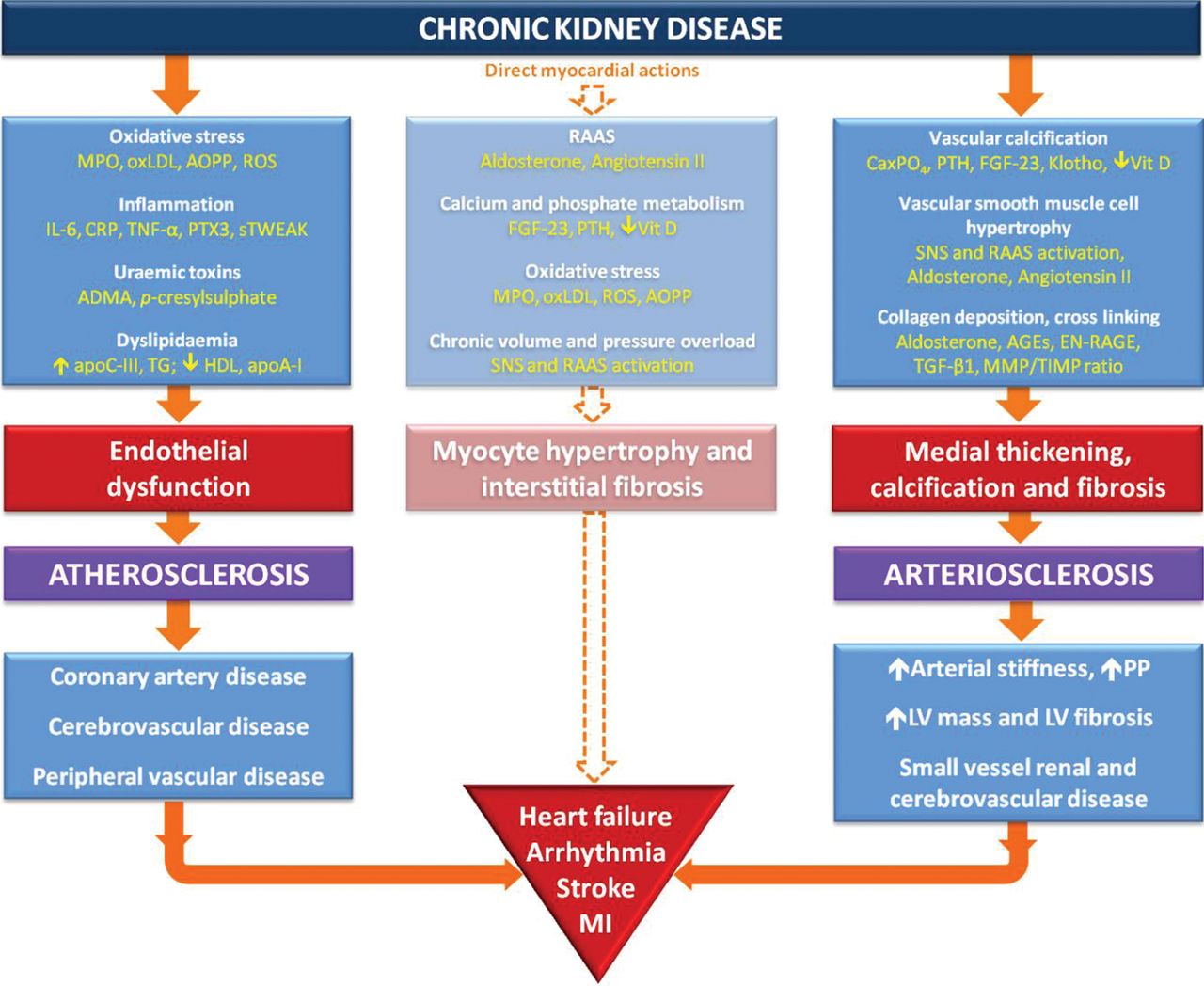
A powerful method of examining the pathogenesis of atheroma in CKD is to measure endothelial function in carefully defined cohorts of patients with CKD. The evidence here has been contradictory. Early studies using flow mediated dilatation and circulating biomarkers suggested that patients with early stage CKD had deranged endothelial function.32–34 More recent work has suggested that this may have been due to the inclusion of patients with other risk factors such as hypertension, vasculitis, smoking and even established vascular disease, and that if these factors are excluded as far as possible, endothelial function is abnormal only in more advanced (stages 4 and 5) CKD.35 This observation is difficult to reconcile with evidence that patients with early stage CKD have many factors known to cause endothelial dysfunction such as inflammation, oxidative stress, increased circulating asymmetrical dimethylarginine and activation of the renin-angiotensin-aldosterone system (recently reviewed elsewhere).36–38 The precise relationship between GFR and endothelial dysfunction remains to be elucidated and many potential mechanisms are under investigation (see online supplementary table 1 and figure 4). Powerful new evidence may become available from longitudinal studies of kidney donors who are free from all major vascular risk factors but who experience a significant fall in GFR after donation.39 Many donors will fall into the category of stage 3 CKD after donation and, while survival data have been reassuring,40 a small adverse cardiovascular influence cannot be excluded because donors are ‘super-selected’ for health and, therefore, have better predicted cardiovascular risk than healthy control populations.
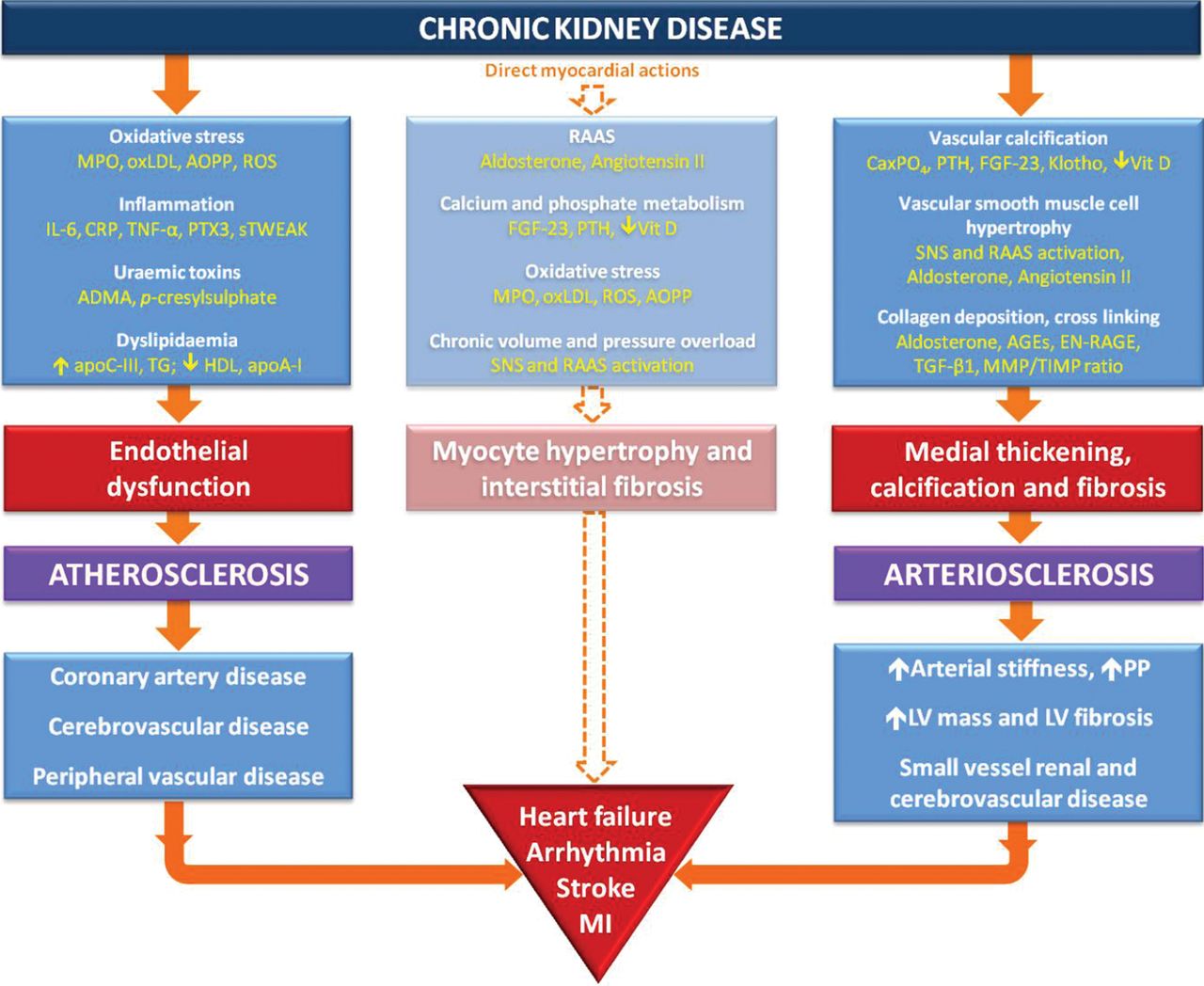
Proposed mechanistic pathways for arterial and myocardial disease in CKD. ADMA, asymmetric dimethylarginine; AGEs, advanced glycation endproducts; AOPP, advanced oxidation protein products; apoA-I, apolipoprotein AI; apoC-III, apolipoprotein CIII; C1TP, carboxy-terminal telopeptide; CaxPO4, calcium phosphate product; CKD, chronic kidney disease; CRP, C reactive protein; EN-RAGE, extracellular newly identified RAGE-binding protein; FGF-23, fibroblast growth factor 23; HDL, high-density lipoprotein; IL-6, interleukin 6; LV, left ventricular; MI, myocardial infarction; MMP, matrix metalloproteinase; MPO, myeloperoxidase; oxLDL, oxidised low-density lipoprotein; PIIINP, procollagen III N-terminal propeptide; PP, pulse pressure; PTH, parathyroid hormone; PTX3, pentraxin-3; RAAS, renin-angiotensin-aldosterone system; ROS, reactive oxygen species; SNS, sympathetic nervous system; sTWEAK, soluble tumour necrosis factor-like weak inducer of apoptosis; TG, triglycerides; TGF-β1, transforming growth factor β1; TIMP, tissue inhibitors of metalloproteinases; TNF-α, tumour necrosis factor α; Vit D, 1,25 dihydroxyvitamin D.
Treatment of atheromatous disease in CKD
There have been few trials examining cardiovascular risk reducing therapies in CKD. Indeed, most trials of such treatments in large populations have excluded patients with significant CKD. Although aspirin appears to be as effective in reducing adverse events after myocardial infarction in patients with ESRD as in those with normal renal function, it may be associated with increased bleeding rates.23 ,41 Its efficacy in patients with CKD and stable coronary artery disease is unknown. The efficacy and safety of antiplatelet therapy in CKD cannot be assumed; a recent systematic review (based on low-quality evidence from post hoc analyses) found that the use of glycoprotein IIb/IIIa inhibitors and thienopyridines in CKD patients with acute coronary syndromes had little or no effect on all-cause or cardiovascular mortality but increased serious bleeding.41 Evidence on the use of statins in CKD is clearer. Although they appeared ineffective in reducing cardiac events and mortality in two small trials of ESRD patients,42 ,43 an analysis of pravastatin use in over 4000 patients with early stage CKD suggested comparable efficacy to non-CKD populations, with a greater absolute effect related to the increased event rate in CKD.44 In SHARP, treatment with simvastatin and ezetimibe was associated with a significant 17% reduction in major atherosclerotic events during almost 5 years of follow-up in patients with ESRD and early stage CKD.21 Treatment with ACE inhibitors is effective in early stage CKD. In PROGRESS (Perindopril pROtection aGainst REcurrent Stroke Study), the risk of recurrent stroke was reduced by perindopril with a larger effect size in patients with stage 3 or greater CKD than that seen in patients without CKD.45 In the HOPE (Heart Outcomes Prevention Evaluation) study, the reduction in death and adverse cardiovascular events in patients with early stage CKD was equal to that observed in patients without CKD.46
Arteriosclerosis in CKD
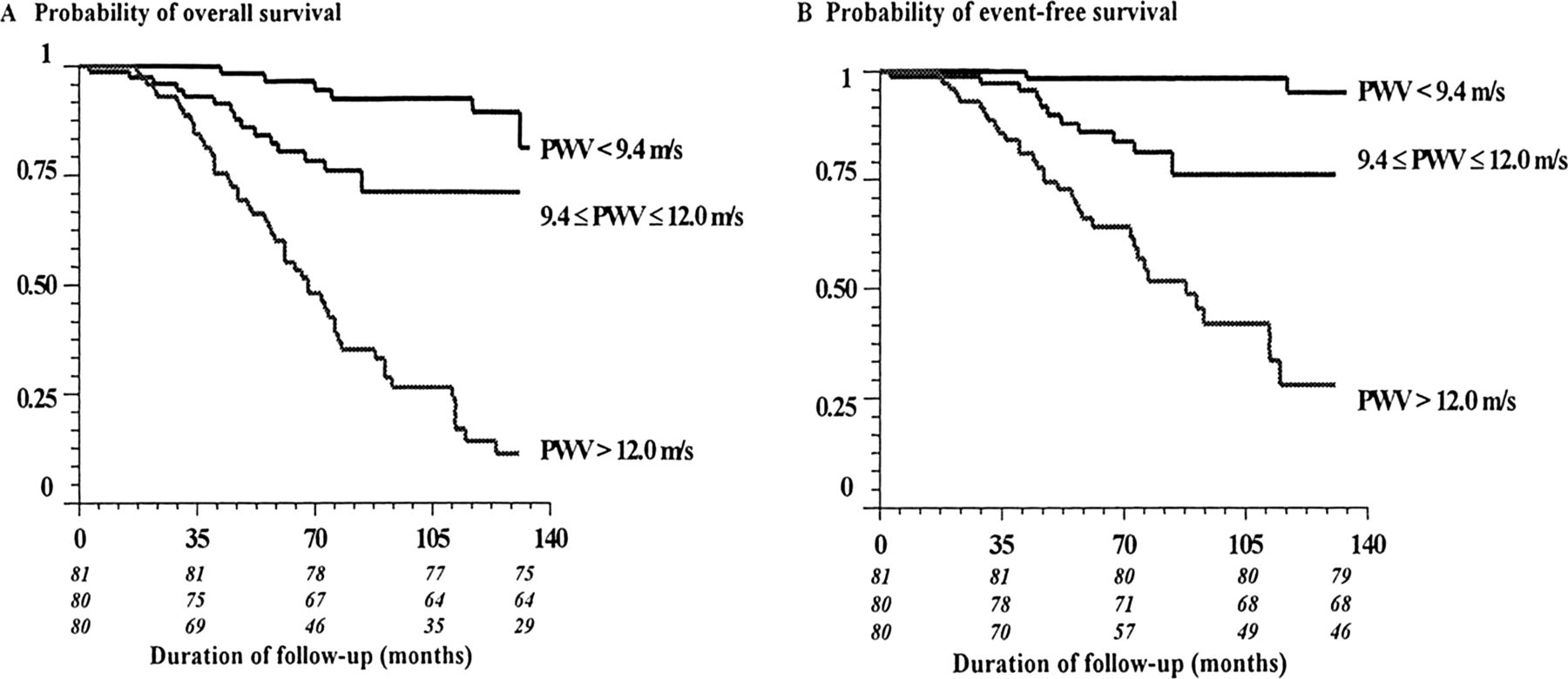
Arteriosclerosis is characterised by thickening and calcification of the medial arterial layer and is a hallmark feature of arterial disease in CKD. Medial calcification is concentric and does not extend into the arterial lumen unless there is coexistent atheroma. Increased collagen content, hyperplasia and hypertrophy of the vascular smooth muscle cells cause wall thickening which in combination with calcification results in increased arterial stiffness.23 Although associations have also been established between the degree of arterial stiffness and atheromatous plaque burden,47 recent studies have failed to demonstrate a significant influence of traditional atherosclerotic risk factors on the development of arteriosclerosis,48 suggesting that alternative factors drive this process. There is certainly some overlap, however, as endothelial dysfunction and reduced NO bioavailability have been shown to contribute to arterial stiffening.49 Increased arterial stiffening appears to plays a central role in the causation of cardiovascular disease in CKD. The strong association between arterial stiffening and mortality in ESRD was demonstrated over 10 years ago by London and colleagues (figure 5).50–52 The process appears to begin during early stage CKD though the prognostic value of arterial stiffness in this group remains unproven.53 ,54
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Probabilities of overall survival (A) and event-free survival (cardiovascular mortality) (B) in ESRD study population according to level of PWV divided into tertiles. Comparisons between survival curves were highly significant (χ2=47.04 for cardiovascular mortality and 67.23 for overall mortality; p<0.0001 for both). Numbers in italics represent individuals at each point according to tertile of PWV. ESRD, end stage renal disease; PWV, pulse wave velocity. From Blacher et al (1999).51
The pathophysiological effects of arteriosclerosis and arterial stiffening are best understood by an appreciation of the normal physiology of the aorta and large arteries. Their major functions are to deliver blood around the body and to buffer the oscillatory changes in BP that result from intermittent ventricular ejection. The highly distensible arterial system ensures that most tissues receive near steady flow with no exposure to peak systolic pressures; this mechanism is so efficient that there is almost no drop in peripheral mean arterial pressure compared with the ascending aorta.55 Loss of arterial distensibility results in a more rigid aorta that is less able to accommodate the volume of blood ejected by the left ventricle, resulting in greater pressure augmentation in systole and higher pulse pressures.56 An often cited additional explanation for the elevated systolic pressure that accompanies an increase in arterial stiffness is that of an earlier return of reflected waves of ventricular contraction from distal arterial branch points.57 ,58 In healthy compliant arteries, reflected waves were believed to return to the ascending aorta in diastole, augmenting diastolic pressure and coronary blood flow; in aged and stiffer arteries, the reflected waves were thought to return earlier in systole increasing ventricular afterload and augmenting systolic and pulse pressures. Although an attractive hypothesis, there is now abundant evidence showing that reflected waves arrive in systole irrespective of age.59 These data together with information available from techniques that separate reflected waves from the reservoir pressure strongly suggest that the ‘cushioning effect’ or Windkessel model appears to be the more important physiological explanation.60 Regardless of the underlying mechanism, as arterial stiffness increases, the loss of arterial distensibility exposes the myocardium, brain and kidneys to higher systolic pressures and greater pressure fluctuations resulting in myocardial, cerebral and renal microvascular damage and an increased risk of heart failure, arrhythmia, stroke and further renal impairment (figure 4).61 While the high systolic pressure increases LV afterload, lower diastolic pressure reduces diastolic coronary perfusion, promoting ischaemia and placing greater reliance on systolic coronary perfusion.62 ,63
Arterial–ventricular interaction in CKD
The central role of arterial stiffening has also focussed attention on the interaction between the left ventricle and arterial tree termed arterial–ventricular interaction. In health, cardiac function is physiologically matched or coupled with arterial function to ensure maximum cardiac work and efficiency.64 This coupling is most commonly investigated and quantified by the measurement of arterial and ventricular elastances, either invasively with conductance catheters or non-invasively using echocardiographic parameters. LV systolic elastance (Ees) is a measure of chamber stiffness at end systole and arterial elastance (Ea) is a measure of net ventricular afterload (incorporating resistance, pulsaltile load and BP) determined by the ratio of end-systolic pressure to stroke volume. When LV function is normal, the coupling ratio is generally between 0.7 and 1.0 (Ea is smaller than Ees) so that work efficiency is maximised but stroke work is submaximal for a given preload. With systolic dysfunction, Ees falls so that in mild dysfunction, the ventricle performs at maximum stroke work. When the heart is coupled to a stiff vascular system, the heart must increase systolic stiffness (Ees) to maintain cardiac metabolic efficiency ensuring transfer of blood to the arterial tree without excessive changes in pressure. Thus, the arterial to ventricular coupling ratio is maintained but both Ea and Ees are considerably elevated. These compensatory adaptations maintain cardiac performance with enhanced contractility at rest, but at a price: cardiac reserve is reduced, diastolic function is impaired and the cardiovascular responses to alterations in pressure and volume load are blunted leading to haemodynamic instability and a susceptibility to ‘flash’ pulmonary oedema.64
The abnormalities of arterial and LV function that are so highly prevalent in ESRD may begin in early stage CKD. In a population-based study of 742 individuals, mildly reduced eGFR was significantly associated with greater LV mass in a process that appeared dependent upon increased arterial stiffness.65 In a cross-sectional study of patients with stages 2 and 3 CKD, we found delayed ventricular relaxation, increased LV systolic and end diastolic stiffness and elevated left atrial volumes compared with healthy matched controls.53 Ea was also elevated such that the arterial to ventricular coupling ratio was preserved. This suggests that the two processes occur in parallel and lends support to the theory that aortic stiffness drives the development of LV stiffness in CKD.53 This pattern, similar to that found in heart failure with preserved ejection fraction,66 suggests that early stage CKD is likely to be associated with reduced cardiovascular reserve and increased haemodynamic instability, although further work is required in this area. The level of GFR at which arterial stiffening begins to increase is not clear but abnormalities in arterial stiffness as determined by pulse wave velocity (PWV) are already evident in stage 2 CKD.67 A stepwise increase in PWV corresponding with advancing stages of CKD suggests that arterial stiffness increases as a graded relationship with declining GFR.
Causes of arteriosclerosis in CKD and implications for treatment
An understanding of the potential mechanisms underlying increased arterial stiffness in CKD is essential for devising strategies to prevent or reverse this pathophysiology. Arterial stiffening is a result of both functional and structural abnormalities of the arterial wall. Endothelial dysfunction provides a dynamic component that may be amenable to treatment with conventional agents such as statins, BP lowering drugs and novel treatments that increase nitric oxide bioavailability. The structural vascular changes of CKD are however likely to prove harder to reverse. The probable causes of arteriosclerosis in CKD have been reviewed in detail in this journal recently68 and are described in the online supplementary table 1. Recent work has highlighted the importance of both aldosterone and disordered bone mineral metabolism in the causation of arterial stiffness in CKD. Preventive therapy is available for both factors; such treatment might reduce arterial stiffness and prevent many of the pathological cardiovascular changes seen in CKD including LV hypertrophy and fibrosis. Reduction of arterial stiffness also has the potential to slow the decline in renal function thereby interrupting a classic vicious circle of renal and vascular disease progression (figure 4).
Aldosterone levels are elevated in CKD as a result of two mechanisms: aldosterone ‘escape’ describes inappropriately elevated plasma levels relative to salt and fluid overload69 while aldosterone ‘breakthrough’ refers to persistently high levels despite treatment with ACE inhibitors and angiotensin receptor blockers.70 It is now well recognised that aldosterone, probably produced locally as well as by the adrenal cortex, exerts injurious actions directly on the myocardium and vasculature. Aldosterone mediated oxidative stress and inflammation result in structural fibrotic change within the arterial wall and myocardium.71 These features may result from both genomic actions via mineralocortcoid receptor (MR) activation and non-genomic- actions. The adverse inflammatory effects of aldosterone are powerfully potentiated and may be dependent upon the presence of sodium excess. The combination of high levels of aldosterone and sodium overload is unusual as aldosterone production is usually suppressed by sodium overload but aldosterone escape is a characteristic of both heart failure (in which MR blocking drugs have proven efficacy) and CKD.69 Aldosterone causes a range of other adverse vascular effects including endothelial dysfunction, autonomic dysfunction, vasoconstriction and hypertension.72 ,73 All of these actions, including inflammation and fibrosis, also occur within the kidney causing further glomerular injury and setting up a toxic feed-forward loop of renal and vascular damage.74 The use of MR blocking drugs prevent many of the adverse effects of aldosterone both in vitro and in animal studies75 suggesting that if these agents can be tolerated without serious hyperkalaemia in patients with CKD, they may be able to prevent or even reverse arterial stiffening and LV hypertrophy and fibrosis. In a preliminary proof of concept study, the effects of spironolactone on arterial stiffness measured by PWV and LV mass measured by cardiac MRI were compared with placebo in patients with stage 2 or 3 CKD.76 After 40 weeks of treatment, significant reductions in both PWV and LV mass were seen in the spironolactone group. Treatment appeared safe with very few adverse effects such as hyperkalaemia.77 Further work is required to confirm safety and to determine whether these results were specific to the MR blockade or might have been secondary to BP reduction.
Serum phosphate is strongly associated with mortality and adverse cardiovascular events in patients on dialysis and in patients with early stage CKD.78 ,79 In healthy individuals and in patients after myocardial infarction, serum phosphate levels even within the normal range are associated with an increased risk of adverse vascular events, higher coronary calcium scores and increased cardiovascular mortality.80–82 It is now evident that CKD mineral bone disorder occurs early in the course of CKD and is associated with cardiovascular disease and increased arterial stiffness.83 ,84 In response to any increase in serum phosphate, parathyroid hormone (PTH) levels rise and fibroblast growth factor 23 (FGF-23) is secreted by osteoblasts and osteocytes. These phosphaturic hormones increase urinary phosphate excretion to maintain phosphate concentrations within the normal range in early stage CKD. Active vitamin D (1,25-dihydroxyvitamin D) synthesis is reduced and is further inhibited by FGF-23 leading to hypocalcaemia and further elevation of PTH, itself a cardiovascular toxin.85 In the advanced stages of CKD, despite massive elevation of FGF-23 and PTH, these regulatory mechanisms fail and phosphate levels begin to rise. Both reduced synthesis of 1,25-dihydroxyvitamin D and increased levels of PTH mediate numerous inflammatory and adverse cardiovascular effects.86 A recent trial examining the effect of an activated vitamin D analogue on LV hypertrophy in stages 3–4 CKD, however, failed to show a reduction in LV mass.87 There is strong evidence that FGF-23 plays a causative role in increasing LV mass in CKD probably by direct effects on the myocardium.88 Klotho, originally identified as an antiageing protein,89 is a transmembrane protein that acts as an obligate coreceptor for FGF-23 in the kidney and parathyroid gland but is not expressed in the heart. Klotho is also cleaved and released into the circulation as a truncated secreted form derived from alternative RNA splicing.90 Recent studies suggest tissue Klotho protects the vasculature from calcification; the role of circulating Klotho is less clear but is emerging as an exciting area of research. Phosphate may also exert direct adverse effects on the myocardium and arterial wall.91 In early stage CKD, phosphate is independently associated with LV mass92 and in vitro work suggests an effect on vascular smooth muscle cells resulting in medial calcification.88
It follows that the use of phosphate binders in CKD has the potential to reduce cardiovascular disease including arterial stiffness. To date, this issue has been difficult to address in clinical studies as many phosphate binders are calcium-based and may exacerbate rather than improve arterial calcification and stiffness. Indeed, the adverse cardiovascular effects of calcium are gaining increasing recognition.93 ,94 Non-calcium-based binders such as sevelamer and lanthanum are an attractive option to reduce arterial stiffness and adverse cardiovascular events in CKD but the results of clinical trials have thus been far been unconvincing, although a reduction in progression of arterial calcification is being demonstrated.95 ,96 A recent small Italian study of 212 patients with CKD stages 3–4 did suggest a lower all-cause mortality in patients randomised to sevelamer compared with patients receiving calcium carbonate.96 Current phosphate binders are poorly tolerated, associated with a high pill burden and exert weak actions on phosphate metabolism, especially in patients with relatively preserved renal function. The hypothesis that reducing phosphate exposure improves cardiovascular outcomes is unlikely to be answered conclusively until newer, more potent and better tolerated agents, such as inhibitors of gastrointestinal phosphate absorption, become available.97
Conclusions
While CKD is associated with a high risk of atherosclerotic disease, arteriosclerosis is the predominant arterial pathophysiology. Medial inflammation, fibrosis, hypertrophy and calcification result in increased arterial stiffness, which accelerates the normal process of vascular ageing, contributing to the development of LV fibrosis and hypertrophy, as well as precipitating further renal damage. Ultimately, this causes high levels of premature mortality due to heart failure and arrhythmias. To date, clinical trials evaluating cardiovascular risk reduction therapy in CKD have proved disappointing; this may be because treatment was aimed at traditional cardiac risk factors which may reduce atheroma burden but have little influence on the progression of arteriosclerosis. Agents aimed at reducing arterial stiffness show greater promise but a better understanding of the many processes causing increased arterial stiffness in CKD is required.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table
Footnotes
-
Contributors All authors were involved in the drafting and critical appraisal of this manuscript.
-
Funding WEM is the holder of a British Heart Foundation Clinical Research Training Fellowship Grant.
-
Competing Interests CDC and CJF have received lecture fees and funding from Genzyme Corporation for an investigator led study.
-
Provenance and peer review Commissioned; internally peer reviewed.