Article Text

Statistics from Altmetric.com
Methotrexate in rheumatoid arthritis
Methotrexate (MTX) is a folate analogue originally synthesised in the 1940s and designed to inhibit dihydrofolate reductase.1 Reduced folate (tetrahydrofolate) is the proximal single carbon donor in several reactions involved in the de novo synthetic pathways for purine and pyrimidine precursors of DNA and RNA required for cell proliferation. Furthermore, tetrahydrofolate plays a part in a second important biochemical step: the methionine-homocysteine cycle, which is necessary to provide a methyl group for several downstream reactions such as methylation of DNA, RNA proteins, and others. Therefore, MTX has been used extensively for treatment of neoplastic diseases. In 1951 the rationale for the introduction of MTX for the treatment of rheumatoid arthritis (RA) was that it inhibited proliferation of the lymphocytes and other cells responsible for inflammation in the joint.2 No further studies on clinical experience with MTX in RA were published until the early 1980s, when several uncontrolled trials were reported.3-8
Finally, four well designed, blinded, placebo controlled studies published in 1984 and 1985 introduced the use of MTX in the treatment of RA.9-12
The early indications for MTX use in the rheumatic diseases were first reported in a large review in 1984.13 From the considerable experience obtained over the past 15 years, several lines of evidence clearly suggest that MTX does not act simply as a cytotoxic (antiproliferative) agent for the cells responsible for the joint inflammation in RA.14 As a matter of fact, it would be difficult to understand how a drug that diminishes inflammation by preventing proliferation of immune cells might work at effective concentrations for only a very short time and once a week. In addition, the rapid clinical remission and the short term effect on the acute phase reactants, as seen with low dose MTX administration in most patients with RA, as well as the fast flare of disease after drug discontinuation, suggest that the mechanism of action of low dose MTX might be more anti-inflammatory than antiproliferative (immunosuppressive).15 ,16
Recently, MTX has been shown to possess a variety of anti-inflammatory effects.17 Although, few studies suggest any specific effect of MTX on T cell number or function in patients with RA, MTX does exert clear inhibitory effects in vivo and in vitro on neutrophils and particularly on monocytes/macrophages that are believed to have a central role in RA pathophysiology and inflammatory synovitis.17-23
These and other separate lines of evidence support the view that alternative mechanisms are responsible for the antirheumatic/anti-inflammatory effects of MTX, which will be reviewed here.
Cellular effects of MTX
MTX is a folate analogue with an amino group (NH2), a methyl group (CH3), and a fully oxidised pteridine ring, rendering the molecule inactive as cofactor.1
Once administered MTX is delivered to cells in the same way as the parenteral folates; 3–12% is hydroxylated in liver and circulates as 7-OH-MTX.24
Extracellular MTX is brought into the cell by the folate receptors (FRα, FRβ). Thereafter, a portion of intracellular MTX and 7-OH-MTX is metabolised to polyglutamates (MTX-glu) in the same manner as naturally occurring folates.25 MTX-glu represent long lived derivatives, which in rats may be detected in the skin for as long as two weeks after a single dose of the drug.26
Because there is a latent period of weeks before the MTX effects are appreciated in patients with RA, it may be the intracellular MTX-glu derivatives which are the true active anti-inflammatory agents.
MTX binds dihydrofolate reductase (DHFR) with high affinity. MTX-glu binds DHFR and has fairly high affinity for enzymes that require folate cofactors, including thymidylate synthetase (TS) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase. The inhibition of TS, induced by MTX, interferes with DNA synthesis in actively dividing cells, and the increase of AICAR enzyme system, which plays a key part in the purine metabolism of the cell, leads to enhanced release of adenosine into the blood.23 ,27 ,28
In fact, a number of anti-inflammatory effects exerted by MTX seem to be related to the extracellular adenosine increase and its interaction with specific cell surface receptors, with subsequent inhibition such as interleukin 8 (IL8) production by peripheral blood mononuclear cells (PBMC), IL6 secretion by human monocytes, leucotriene B4 synthesis in neutrophils, and decreased synovial collagenase gene expression.14 ,29
MTX effects via adenosine induced immunosuppression
MTX typically blocks tetrahydrofolate dependent steps in cell metabolism. Because tetrahydrofolate and polyglutamyl derivatives of tetrahydrofolate are involved in purine biosynthesis several consequences can appear which result in adenosine overproduction. In purine biosynthesis two steps are tetrahydrofolate dependent (fig 1A). There is a preponderance of MTX mediated inhibition of the second enzyme AICAR formyltransferase in comparison with the first enzyme GAR formyltransferase (fig 1A).27 ,30

Adenosine increase by MTX and subsequent immunosuppression through adenosine receptors. (A) MTX inhibits both, conversion of GAR → FGAR and AICAR → FAICAR. However, inhibition of the second step is stronger, which results in accumulation of AICAR. (B) Accumulated AICAR inhibits AMP deaminase and adenosine deaminase (ADA), which increases adenosine-5′-P and adenosine (C). (D) Intracellular accumulation of adenosine-5′-P and adenosine results in an increase of these compounds in the extracellular space. Here, adenosine-5′-P is converted to adenosine, which binds to the specific receptor subtypes A1, A2a, and A2b (E). Probably, there will be a preponderance of the A2 receptor pathway, yielding an increase of cyclic adenosine monophosphate (cAMP) in the cell (F). (G) cAMP increase leads to immunosuppression. AMP = adenosine-5′-monophosphate; MTX = methotrexate.
Thus there will be a relative increase of AICAR (fig 1A). AICAR itself inhibits important steps of degradation of adenosine-5′-P and adenosine by the AMP deaminase and the adensosine deaminase (ADA), respectively (fig 1B).
Inhibition of degradation of these two intracellular compounds leads to increased intracellular (fig 1C) and extracellular adenosine-5′-P and adenosine (fig 1D).27 At the surface of different types of immune competent cells, the ecto-5′-nucleotidase (CD73) converts adenosine-5′-P to adenosine (fig 1D).31 ,32 This surface enzyme can be regulated by several immune mediators, such as interleukin 4 (IL4) and interferon γ (IFNγ), which decrease the activity of the ecto-5′-nucleotidase on PBMC.33 ,34 On the other hand, it has been reported that IL1 and tumour necrosis factor (TNF) increase activity of this enzyme.35 Thus the local microenvironment determines the activity of this enzyme.
Extracellular adenosine can bind to the seven transmembrane-spanning adenosine surface receptors types A1, A2α, A2β, A3, which have been found on many different cell types (fig 1E).17
The rank order of the affinity of adenosine binding to these receptor subtypes is A1>A2a>A2b.36 The adenosine A1 receptor is coupled to a Gαi/o protein and the A2a and A2b receptors are coupled to GαS (fig 1). Ligation of A1 receptors decreases intracellular cyclic adenosine monophosphate (cAMP) (fig 1), whereas binding of adenosine to A2 receptors increases intracellular cAMP (fig 1). If the pathways through the two different receptor subtypes A1 or A2a/b were functionally intact one would expect a preponderance of the A1 pathway owing to the higher affinity of adenosine to the A1 receptor subtype. This would lead to a decrease of cAMP (fig 1). However, low dose MTX exerts its anti-inflammatory effect by inducing extracellular adenosine, which acts predominantly through A2a receptors.14 ,17 ,37 ,38
Thus it seems as if A1 receptor signalling is switched off. Similar effects have been described in a proinflammatory situation where the pathway through the two receptors is shifted to GαS (fig 1F), yielding an increase of cAMP.39
Furthermore, it has been shown that cytokines can up regulate the A2 receptor subtype, which may be another mechanism to shift the pathways to GαS rather than Gαi/o.40 ,41 It has been repeatedly shown that an increase of cAMP leads to immunosuppression by inhibition of phagocytosis, inhibition of secretion of TNF, IFNγ, IL2, IL12, HLA expression, and many others.42-47
For adenosine via A2 receptor binding it has been specifically shown that this substance inhibits lymphocyte proliferation and production of TNF, IL8, and IL12.29 ,48 ,49 On the other hand, adenosine via A2 receptor binding increases secretion of IL6 and IL10.50 ,51 Binding of adenosine to A3 receptors leads to inhibition of secretion of TNF, IL12, and IFNγ.52 ,53 In conclusion, binding of adenosine to A2 and A3 receptors results in a favourable situation which is probably one of the important anti-inflammatory mechanisms of MTX action.
MTX effects on immune/inflammatory cell proliferation and apoptosis
Recent data have already suggested that the disruption of the cell cycle caused by high dose MTX treatment may be the initial step of the apoptotic sequence of dying cells and may explain the antiproliferative effects of the drug.54 The involvement of the APO-1/Fas (CD95) receptor/ligand system in MTX induced apoptosis has been recently identified in leukaemia cells, with a peak of apoptosis between 24 and 48 hours.55
In addition, MTX was found to inhibit markedly the spontaneous proliferation of U937 monoblastic leukaemia cells in vitro and induce the rapid expression of the apoptosis receptor CD95 also in presence of 1,25-OH-cholecalciferol.56 Results of another recent investigation, are in agreement with these latter studies and seem to suggest that intermediate MTX concentrations (50 μg/ml), as obtained in serum after low dose treatment, can induce both a significant cell growth inhibition and apoptosis, at least in monocytic immature cells (THP-1 cell line)57 (fig 2A). For cell proliferation, the lowest in vitro MTX concentrations (from 5 to 500 ng/ml) were confirmed to be ineffective.54 In that study no significant effects on synovial macrophage proliferation were obtained with an MTX concentration of 50 μg/ml (achievable in the serum with low dose MTX treatment in RA).57

{kind=link}
{kind=link}
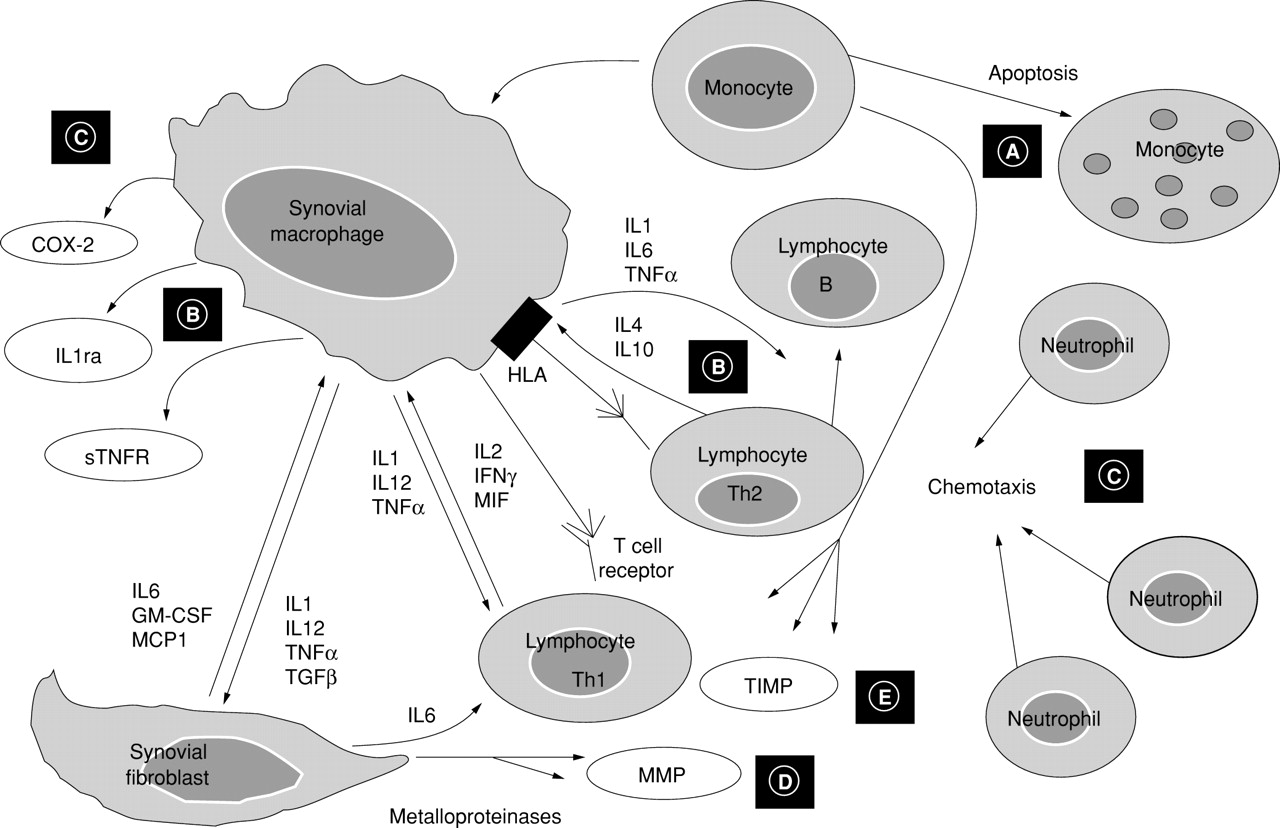
Anti-inflammatory effects exerted by low dose MTX at the level of the synovial tissue in RA. (A) MTX reduces monocytic cell growth and increases their apoptosis. (B) MTX decreases the IL1 and IL6 secretion and increases IL1ra production. At the same time, MTX increases IL4 and IL10 gene expression and decreases gene expression of proinflammatory Th1 cytokines (IL2 and IFNγ). (C) MTX seems to exert indirect inhibition of COX-2 synthesis and neutrophil chemotaxis. (D) MTX exerts indirect inhibitory effects (through modulation of cytokines) on synovial metalloproteinase (MMP) production and stimulates their inhibitors (TIMP) (E). MTX = methotrexate; IL1ra = interleukin-1 receptor antagonist; IFNγ = interferon γ; COX-2 = cyclo-oxygenase-2; MMP = metalloproteinase; TIMP = tissue inhibitor of metalloproteinase.
The explanation for the lack of modulatory in vitro potency of MTX on synovial macrophage growth and apoptosis, as already found for cyclosporin A, may be that MTX affects only immature differentiating monocytes and not differentiated cells (that is, tissue infiltrating monocytes and resident macrophages, respectively).58 ,59Therefore, these findings suggest that MTX might inhibit recruitment of immature and inflammatory monocytes into inflammatory sites and could reduce the survival of these cells in the inflamed synovial tissue.57
A recent paper investigated whether other immunosuppressive properties of low dose MTX treatment were related to apoptosis.18
The study showed that activated T cells from human peripheral blood underwent MTX induced apoptosis, which was completely abrogated by addition of folinic acid. Apoptosis of activated T cells did not required interaction between CD95 (APO-1/Fas) and its ligand, and adenosine release accounted for only a small part of this MTX activity. Finally, in vitro activation of peripheral blood taken from patients with RA after MTX injection resulted in apoptosis.18
However, several studies have recently shown that low dose MTX may well induce antiproliferative effects on immune cells owing to inhibition of dihydrofolate reductase and folate dependent transmethylations as apoptosis independent mechanisms.
A recent study showed that patients with RA, treated with MTX, expressed low concentrations of circulating purines and pyrimidines, with consequent reduced availability for DNA and RNA synthesis and cell proliferation.60
Inhibition of mononucleotide precursors of nucleic acid, particularly at the step of methylation of dUMP into dTMP by thymidylate synthase, represents a further cause of disruption of DNA synthesis and inhibition of proliferation of cells involved in the inflammatory process in the joints.
A recent paper confirmed that low concentrations of MTX inhibited in vitro thymidylate synthase activity in human PBMC.61
As initially discussed, it is difficult to relate observed changes in purine and pyrimidine levels directly to the pharmacokinetics of MTX because MTX clearance from blood is rapid. Therefore, metabolic effects of MTX could be attributed predominantly to its polyglutamated derivatives which are formed and accumulate inside the cells. MTX polyglutamate derivatives may interfere with purine and pyrimidine metabolism and explain long term antiproliferative effects in RA after low dose treatment with MTX once a week.62
In conclusion, the anti-inflammatory effects of MTX on cells responsible for joint inflammation in RA might be, at least partially, linked to antiproliferative and apoptosis related mechanisms.
MTX effects on monocytic and lymphocytic cytokines and their inhibitors
By considering IL1 and TNFα, which are cytokines with a central role in the inflammatory process and which are mainly produced by monocytes/macrophages at the level of the RA synovial tissue, early studies suggested that MTX inhibits IL1 production in vivo and ex vivo.20 ,63
More recently, it has been suggested that MTX interferes directly with the binding of IL1 to its receptor and thereby inhibits the cellular responses to IL1.64 Alternatively, MTX, through adenosine increase and binding to A3 receptors, might promote the IL1 receptor antagonist (IL1ra) transcription and presumably its production. Recent studies seem to confirm this possibility, because it has been shown that MTX treatment generates a less inflammatory type of circulating monocyte in patients with RA treated with low doses, by inhibiting IL1 and IL8 secretion and, in parallel, by inducing the IL1ra (fig2B).65 ,66
A more recent study seems to confirm the possible IL1ra mediated anti-inflammatory effects of MTX, at least on monocytes, because a significant increase of IL1ra was found with the low dose treatment of human cultured monocytic THP-1 cells.57
The effects were clearly dose dependent and gradually decreased with the lowest MTX concentrations at 24 hours, while the presence of a steady state (at 48, 72, and 96 hours) indicated also time dependent effects. Interestingly, the high doses of MTX were found to cause a significant IL1ra decrease on cultured THP-1 cells compared with untreated control cells; this decrease was probably due to cell damage (apoptosis).57
Monocytes produce greater amounts of IL1 than IL1ra, whereas macrophages produce mainly IL1ra in in vitro cultures.67Recent results confirm these data, because IL1ra basal production was found to be significantly higher from untreated synovial RA macrophages than from untreated monocytic THP-1 cells.57
In fact, the maturation into macrophages of monocytes that enter RA synovial tissue is characterised by different phenotypic and functional changes that seem to include higher production of IL1ra.68 ,69
An excess of IL1ra production occurs in active RA, as shown by the very high levels of the protein found in the synovial fluid of patients with RA, even though IL1ra production in the RA synovial tissue may not be in sufficient excess to inhibit the proinflammatory effects of locally produced IL1.70
The previously reported study showed a significant increase of IL1ra with low dose MTX treatment of cultured monocytic THP-1 cells, whereas MTX was unable to change IL1ra levels produced by cultured RA synovial macrophages.57
Recently, it has been also shown that adenosine inhibits TNFα expression in a monocytic cell line and that monocytes release adenosine after treatment with MTX.52 ,71
In addition, recent investigations showed both a late up regulation of the soluble TNFα receptor (sTNFR p75) synthesis by PBMC after 24 hours of MTX treatment and the MTX induced increase of sTNFR p75 from cultured monoblastic leukaemia cells, suggesting a further anti-inflammatory mechanism through inhibition of TNFα effects.56
The short term anti-inflammatory effects of MTX may include the inhibition of IL6 secretion by cultured human monocytes and, in the course of RA treatment, a decreased production of IL6, which might correlate with improvement of biological parameters of disease activity.29 ,72 ,73
It has proved difficult to study cytokines of mainly lymphocytic origin in RA, such as IL4, IL10, IL2, and IFNγ, because they are only weakly expressed and produced in the RA synovial tissue. However, in a recent study using the gene amplification by reverse transcriptase-polymerase chain reaction the effects of MTX on gene expression for these cytokines were analysed in PBMC of patients with RA.74 The study showed that under the action of MTX in vitro, there was increased IL4 gene expression by phytohaemagglutinin stimulated PBMC from patients with RA. In addition, MTX increased IL10 gene expression in the same cells. The increased expression of IL4 and IL10, two cooperative cytokines with anti-inflammatory properties, may further partly explain the efficacy of MTX in RA (fig 2B).75Moreover, the action of MTX decreased expression of IL2 and the IFNγ gene in the PBMC of patients with RA.75
This decreased MTX induced expression of IL2 and IFNγ genes (phenotype Th2) may be secondary to the action of IL4 and IL10, which inhibit the activity of lymphocytes of the Th1 phenotype.76
In conclusion, MTX treatment in RA seems to reduce the production of proinflammatory monocytic/macrophagic cytokines (IL1, IL6, and TNFα), to increase, at least, gene expression of anti-inflammatory Th2 cytokines (IL4 and IL10), and to decrease gene expression of proinflammatory Th1 cytokines (IL2 and IFNγ), with resulting anti-inflammatory effects.
MTX effects on cyclo-oxygenases and lipoxygenase
Prostaglandins and leucotrienes are strongly involved in the inflammatory reaction. In particular, prostaglandins are important mediators of joint destruction in RA. A recent study investigated the effects of MTX on cyclo-oxygenase (COX) metabolism by evaluating the prostaglandin E2 (PGE2) synthesis in cultured human rheumatoid synoviocytes.77 The results showed a dose dependent decrease of IL1 induced PGE2production by cultured RA synoviocytes that was determined by MTX treatment, whereas neither COX-1 nor COX-2 mRNA expression was affected by MTX incubation.77
In a more recent study the effects of MTX on COX-1 (thromboxane B2) and COX-2 (PGE2) activity were evaluated in whole blood of patients with RA treated with MTX.78Interestingly, COX-2 activity was found to be reduced in the plasma of patients with RA treated with MTX in comparison with healthy controls. Inhibition of COX-2 activity was also found when blood of normal donors was co-incubated with the serum of MTX treated patients with RA. However, direct action of MTX on either enzyme was excluded.78
The inhibitory effect exerted by MTX on neutrophil chemotaxis, found in synovial fluid of patients with RA, might further determine decreased COX concentration in inflammatory joints (fig 2C).79
Leucotriene LTB4 is a 5-lipoxygenase product that might also stimulate IL2 and IFNγ production by T cells. MTX has been found to decrease both the synthesis of LTB4 by neutrophils and total plasma LTB4 concentration in patients with RA treated weekly with 10 mg of MTX.80 Possible pharmacokinetics and clinical problems after the coadministration of MTX, particularly at a high weekly maintenance dose (but not at 7.5 mg), and non-steroidal anti-inflammatory drugs, have been suggested/observed in relation to their competition for renal tubular excretion or impairment of hepatic metabolism.81 ,82
However, a recent study found no significant effect on MTX pharmacokinetics in patients with RA, by evaluating coadministration of MTX and a specific COX-2 inhibitor.83 In conclusion, the mostly indirect inhibition exerted by low dose MTX on cyclo-oxygenases and lipoxygenase products might well account for its anti-inflammatory action seen in RA.
MTX effects on metalloproteinases and their inhibitors
The destruction of joints in RA is thought to be related in part to the increased synthesis and activity of proteolytic enzymes released by inflammation activated cells and represents a further cause of chronic inflammation.
An early study, showed that MTX markedly decreases the neutral metallocollagenolytic enzyme (NMCE) activity, as well as synovial and cartilage tissue NMCE levels in patients with RA treated with MTX, in comparison with untreated controls.84
In addition, a retrospective analysis of specimens obtained at the time of joint replacement surgery, indicated that synovial tissue from patients with RA treated with MTX had less fibrosis than did that from patients treated with other drugs, further suggesting an MTX effect on decreasing proteinase production.85
A recent study analysed, by in situ hybridisation, the messenger RNA (mRNA) levels of collagenase, stromelysin, and tissue inhibitor of metalloproteinase-1 (TIMP-1) on frozen synovial tissue sections of patients with RA before and after treatment with low dose MTX (fig2D).86 The results showed a significant decrease of the collagenase gene expression after MTX treatment, whereas TIMP-1 and stromelysin mRNA levels were unchanged. In addition, MTX did not alter collagenase or TIMP-1 mRNA levels on MTX treated fibroblast-like synoviocytes after IL1 exposure.86
One might argue that proteinase reduction is probably caused by simultaneous MTX mediated down regulation of IL1, which induces proteinases in synovial fibroblasts87 (fig 2E). Therefore, cytokine regulation seems a likely direct cause of proteinase gene modulation, and the MTX effects seen on metalloproteinases and their inhibitor levels were suggested to represent an indirect effect rather than a direct influence on gene expression, owing to an MTX related alteration of the synovial cytokine milieu.86 ,88 A more recent investigation confirmed the effects of low dose MTX treatment on metalloproteinase-1 (MMP-1) and TIMP-1 levels, by analysing at baseline and after four months, synovial tissue samples obtained from patients with RA.79
In this study, once again a significant decrease of the MMP-1/TIMP-1 ratio was found in the synovial tissue of MTX treated patients; the decrease was due to reduction of the MMP-1 levels while TIMP-1 levels were found to be relatively unchanged (fig 2E).79
Finally, another recent analysis showed, both ex vivo and in vitro, that enhanced TIMP-1 production by PBMC of patients with RA and healthy subjects upon MTX treatment was associated with simultaneously enhanced IL6 release.89 These latter results seem to confirm that direct cytokine regulation by MTX is probably the indirect cause of gene modulation of proteinases and their inhibitors observed in patients with RA after MTX treatment.86
Perspectives and conclusions
Low dose MTX in RA treatment seems to exert anti-inflammatory effects by acting at different levels of the pathophysiological cascade. The direct inhibitory effects on proliferation and the induction of apoptosis in cells involved in the immune/inflammatory reaction, represent undoubtedly the first step of the intervention.
However, the reported inhibition of both monocytic/lymphocytic proinflammatory cytokines involved in rheumatoid synovitis, seems to be the key role in the sustained anti-inflammatory actions exerted by low dose MTX.
In fact, both the decrease in cyclo-oxygenase/lipoxygenase products and the proteinase/TIMP-1 ratio decrease seem to be indirectly related to the earlier cytokine regulation determined by MTX treatment of patients with RA.
Knowledge about the basic mechanisms of action of MTX might help to explain the problem of patients with RA who are non-responders or resistant to treatment. A recent study showed that the folate receptor β (FRβ) expression is selectively increased in RA synovial macrophages and suggests that MTX is transported within the cell through the FRβ.90
However, this in vitro study and another more recent in vivo study both showed that the coadministration of MTX and folic acid reduces the cellular uptake and increases the total clearance of MTX, respectively.91 In addition, a recent study considering some clinical variables showed that patients with RA treated with MTX without folic acid supplementation had significantly lower disease activity than controls treated with both MTX and folic acid together.92 However, the authors concluded that the addition of folic acid to MTX prevented some side effects, even if with a little loss of efficacy. Therefore, as already suspected, the coadministration of MTX and folic acid might represent a possible cause of MTX resistance in RA treatment and seems to be related to their competition for absorption.93 Most studies suggest delaying the administration of folates to avoid interference with the anti-inflammatory effects of MTX.93-96
Studies indicate that adenosine is responsible for the anti-inflammatory actions of MTX, but a recent investigation of the adjuvant arthritis model of RA has shown a reverse of the anti-inflammatory effects using adenosine receptor antagonists, such as theophylline and caffeine.97 In fact, high use of caffeine or theophylline might represent a further cause of reduced response to low dose MTX in RA, as recently confirmed.98
In conclusion, anti-inflammatory effects of low dose MTX treatment in RA, represent a biological and clinical reality.99 The anti-inflammatory activity of MTX represents further important support for its long term use, in particular when combination treatment with other antirheumatic drugs is planned.