Article Text

Statistics from Altmetric.com
- clinical genetics
- aortic aneurysm
- familial cardiomyopathies
- cardiac arrhythmias and resuscitation science
Learning objectives
To integrate the family history and directed genetic testing into the diagnostic evaluation of patients with cardiovascular disease.
To manage patients and families with inherited cardiovascular conditions.
To manage the uncertainties associated with genetic testing.
To integrate new information about genetic conditions into clinical care.
To recognise phenotypes associated with inherited cardiovascular conditions.
Genetic factors in cardiovascular disease
Over the past decade, there has been increased recognition of genetic causes for many types of cardiovascular disease, with significant implications for patient management depending on the specific genetic condition. Although polygenetic associations with atherosclerotic cardiovascular disease have long been known, a number of inherited single-gene variants resulting in unique cardiovascular phenotypes has been recognised only more recently. Previously, it was difficult to make a genetic diagnosis because testing was expensive and rarely available. The current era of massively parallel DNA sequencing and large volume commercial labs has resulted in wider availability of gene panels and even exome and genome sequencing for use in cardiovascular genetics. Dozens to hundreds of genes of interest can be sequenced at lower cost, far more rapidly than a decade ago, making clinical testing more accessible than ever before.
Many cardiology practice guidelines now incorporate genetic data in recommendations for diagnosis and personalised clinical management. Thus, clinical cardiologists need to understand the basic principles of cardiovascular genetics, recognise which patients might have an underlying genetic condition and refer patients for genetic testing when appropriate. Patients and families with inherited cardiovascular conditions also will expect their physicians to engage in informed shared decision making about clinical management. In this review article, we summarise basic principles of medical genetics and provide a practical approach to clinical genetic testing focusing on three categories of cardiovascular diseases: aortopathies, cardiomyopathies and arrhythmias.
Genotype versus phenotype: one or many genes?
Genetic variations in patients are broadly divided into two categories: benign common nucleotide variations versus pathogenic gene variants (eg, mutations) that result in clinical disease.
Single nucleotide polymorphisms (SNPs)
SNPs (or ‘snips’) are non-pathogenic variations at specific locations in the DNA nucleotide sequence with a population frequency at least 1%. Each person has about 4–5 million SNPs; most are found in non-coding regions of the DNA and have no known effects on health. However, some SNPs occur in regulatory regions of the DNA or occur within a gene and may predict risks of specific diseases or responses to medications and environmental factors. Typically, the association of specific SNPs with cardiovascular diseases represent an additional risk factor in a multifactorial disease process rather than being due to coding for an abnormal protein.
Mendelian randomisation studies take advantage of the random allocation of SNPs in a population to study the causal effect of specific biomarkers on disease.1 For example, in a large population-based study, Mendelian randomisation (based on a group of SNPs associated with systolic blood pressure) was used to show that chronic hypertension increases the risk of aortic valve stenosis by more than threefold for every 20 mm Hg increment in systolic blood pressure.2
Genome-wide association studies (GWAS) allow identification of SNPs associated with cardiovascular disease and may identify pathogenic variants (PVs) by comparing frequencies of thousands of SNPs in patients with a specific disease compared with those without the disease. Advanced statistical approaches are needed given the large number of SNPs examined to ensure that apparent associations are not spurious. Identification of an SNP in a gene encoding a protein that seems biologically plausible as being relevant to the disease process, followed by additional studies showing altered levels of that protein in patients with the disease, lends further support to a pathogenic role of that gene in the disease process. An example of a GWAS study is the association of variation in the lipoprotein(a) (Lp(a)) locus (rs10455872) with calcific aortic valve disease,3 which was further validated in subsequent studies on the association of Lp(a) levels and valve calcification.
Pathogenic variants
PVs (also called ‘mutations’) are a single nucleotide change, deletion or insertion that results in inadequate production or altered function of the protein encoded by that gene, leading to clinical manifestations in most individuals. Examples of a PV resulting in a genetic disease include Marfan syndrome (fibrillin gene) and hypertrophic cardiomyopathy (HCM) (sarcomere genes)4 5 (figure 1). PVs related to cardiovascular disease primarily are germline variants, affecting all cells in the body, unlike the somatic cell variants seen in some types of cancer. A PV usually is inherited from one or both parents; however, new (eg, de novo) mutations also occur in some patients. Autosomal dominant genetic conditions result in phenotypic features with just one copy of the abnormal gene (one allele), whereas autosomal recessive conditions are evident only when both alleles are abnormal. The clinical effects of PVs may be due to inadequate production of the normal protein by the remaining normal allele (haploinsufficiency) or due to the abnormal protein interfering with normal cellular or tissue integrity and function (dominant-negative effect).6 However, the genetics of clinical phenotypes due to a single gene defect are complex with patients having many different specific variations in the affected gene. Even with the same genetic variant, there can be differences between individuals in phenotypic features, a phenomenon called variable expression.
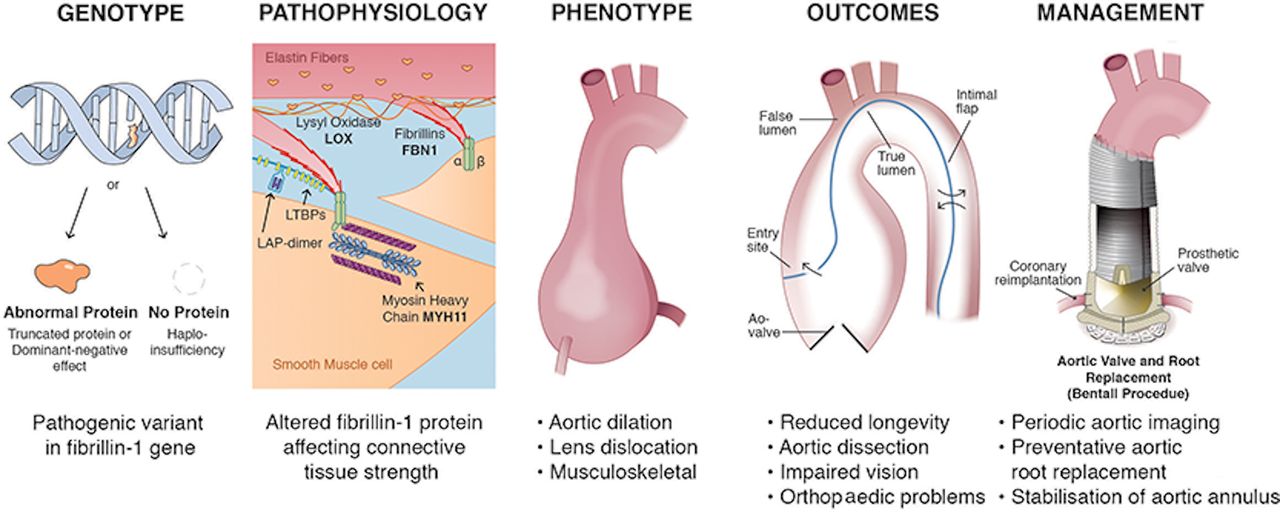
Example of the relationship between genotype, phenotype, clinical outcomes and patient management. In patients with Marfan syndrome, a pathological variant in the gene encoding for the protein fibrillin results in altered strength of the aortic connective tissue. The key phenotypic features are dilation of the aortic sinuses with loss of the normal contour of the sinotubular junction, superior dislocation of the lens in the eye and musculoskeletal involvement. There is a high risk of aortic dissection with current guideline recommending prophylactic replacement of the aortic sinuses and ascending aorta when the diameter reaches 50 mm in asymptomatic patients with Marfan syndrome, or sooner if there is a family history or early dissection or evidence for rapid progression. The surgical procedure includes replacement of the aortic sinuses with coronary artery reimplantation and stabilisation of the annulus. In some patients, the native aortic valve can be reimplanted within the aortic graft but most require prosthetic aortic valve replacement.
Chromosomal abnormalities
Chromosomal abnormalities also are associated with cardiovascular disease but typically have prominent non-cardiac features and usually are diagnosed in childhood because multiple genes are affected. Chromosomal abnormalities associated with cardiovascular disease include the presence of an additional chromosome, such as trisomy 21, which results in complex congenital heart disease7; an entirely missing chromosomes, such as Turner syndrome (monosomy X), which is associated with presence of a bicuspid aortic valve and aortopathy;8 9 or the absence of a chromosome segment, such as diGeorge syndrome (deletion on chromosome 22q11), which is associated with tetralogy of Fallot, truncus arteriosus and ventricular septal defect.10
Clinical genetic testing: whom, what and when?
Whom to test
A core tenet of clinical genetics is to begin genetic testing in a clearly affected patient (the index case or proband) to maximise the likelihood of identifying a PV.11 When more than one family member is affected, test the patient whose onset of disease was at a younger age or who lacks confounding environmental factors that could result a similar clinical presentation. When the index case is identified at autopsy, a postmortem blood or tissue sample can be obtained for DNA analysis.
If no affected patient is available for testing, cardiac phenotyping of first-degree relatives with imaging, ECG, exercise stress testing and so on may be reasonable when there is a family history of a potential inherited cardiogenetic condition. If cardiac phenotyping is normal in relatives, but there is concern for a hereditary aetiology in the deceased, then the limitations of genetic testing of unaffected family members should be clearly understood by the clinician and family.
If genetic testing identifies a PV that fits the clinical features, then cascade testing of family members is recommended. Testing of both parents can identify the side of the family at risk. If both parents test negative, then the PV may have occurred de novo in the proband. Gonadal mosaicism in an unaffected parent (who has negative or low percentage of pathogenic alleles in blood), but their sperm or egg cells harbour the PV, can result in multiple affected offspring. Therefore, genetic testing is offered to siblings even if the parents’ testing is negative.
Each child of a patient with an autosomal dominantly inherited cardiac disease has a 50% risk to have inherited the pathogenic allele and a 50% risk to have inherited the normal allele. A sibling or child who tests negative for a known familial PV is not at risk to develop the disease, does not need continued cardiac evaluation and cannot pass on the disease to children.
What to test
If a PV has been identified in a family member, then it is essential to obtain and review the genetics report for confirmation. If a gene panel was performed and a single PV was identified, then cascade testing for family members should be limited to the single PV. If genetic testing was not done or was non-diagnostic, cardiac genetic testing is most efficiently and economically done by ordering a multigene panel.12 The genes included on a specific panel (eg, cardiomyopathy or aortopathy) vary from lab to lab, the number of genes varies over time and whether testing includes deletion/duplication analysis are points to consider. Consultation with a clinical geneticist or genetic counsellor is recommended for ordering large panels or exome sequencing.13 14 Pretest utilisation review of genetic test orders by a lab genetic counsellor has been shown to reduce the proportion of inappropriate tests by 26%.15
When to test
Some of the common indications for cardiac genetic evaluation include: cardiomyopathies, sudden cardiac arrest in a young person, inherited arrhythmias, muscular dystrophies or Friedreich ataxia associated with cardiomyopathy, aortopathies, congenital heart disease and heritable lipid disorders.11 Younger age of onset and a positive family history increase the pretest probability of identifying a genetic aetiology (figure 2).

Clinical approach to cardiogenetics. Cardiogenetic evaluation is appropriate in patients with a cardiac phenotype suggestive of a genetic cause, a family history consistent with an inherited condition and in patients with a first-degree relative with genetic testing showing a pathogenic or likely PV. The first step is a careful phenotypic evaluation. Genetic testing with a panel of genes or consultation with medical genetics is appropriate in many patients, which determines subsequent medical care for the patient and testing of family members. PV, pathogenic variant.
Phenotypic features
Syndromic genetic conditions comprise features in multiple organ system and are often diagnosed in childhood. In adults, phenotypic features suggesting a genetic condition may be based on clinical history, physical examination or imaging findings. For example, features in some patients with Marfan syndrome include tall stature, arachnodactyly and superior dislocation of the eye lens. Patients with Loeys-Dietz syndrome may have hypertelorism and a bifid uvula. On imaging, the shape of the aortic sinuses and sinotubular junction typically are quite different for a genetic aortopathy compared with hypertensive vascular disease.
Family history
Obtaining a three-generation family pedigree is a standard part of genetic evaluation and can provide important clues to the diagnosis, as well as identify which family members are at risk. Paediatric onset cardiovascular genetic disorders may be inherited as autosomal recessive, autosomal dominant, X-linked or mitochondrial traits. Adult onset cardiovascular genetic disorders are usually inherited in an autosomal dominant pattern (figure 3).

Examples of family pedigrees. (A) X-linked inheritance. Affected males are connected by carrier females. Heterozygous females can be unaffected, mildly affected due to the presence of a second normal X-chromosome or severely affected because of skewed X-inactivation. (B) Autosomal recessive inheritance. Affects both males and females in a single generation; parents are unaffected carriers. (C) Autosomal dominant inheritance. Affects both males and females in multiple generations. Male-to-male transmission exclude X-linked inheritance. Note that individual II-1 is unaffected, but an obligate heterozygote for the familial PV, since he inherited it from his father (I-1) and transmitted it to his son (III-2). This is an example of incomplete penetrance. Square=male; circle=female; filled symbols=affected; dot=carrier; unfilled symbol=unaffected,.
Clinical events
Unfortunately, many patients with a genetic condition first come to medical attention after an acute clinical event such as resuscitated cardiac arrest, aortic dissection or acute heart failure. Other cardiovascular events that are potentially ‘red flags’ for an underlying genetic condition include: carotid artery dissection, syncope during exercise or structural abnormalities found on imaging or at autopsy.16
Genetic testing results: what does the report mean?
Genetic testing results are provided as clinical reports that can be uploaded into the medical record. The American College of Medical Genetics and Genomics guidelines ensure standardisation in reporting across clinical laboratories. The genetic testing report results section contains a list of variants identified, including gene name, nomenclature at the nucleotide and protein level, associated diseases, pattern of inheritance, exon, zygosity and variant classification. The interpretation section provides supporting data for variant classification, predicted effect of the genetic variant on the protein and any further recommendations for clinical testing.
Currently, variants are classified into five categories: benign, likely benign, variant of unknown significance, likely pathogenic and pathogenic. Variant classification is based on data from animal studies, large human databases and segregation of the variant in affected individuals with the disease. Computational analysis of a variant, with modelling of the expected effects of the gene variant on proteins structures and function, also can provide supporting evidence for establishing pathogenicity. Identification of a variant with a high frequency in a human exome database is strong evidence for a benign or likely benign classification. In contrast, a variant likely to cause abnormal function in a gene known to cause disease is strong evidence for a PV or likely PV, which are much less common than benign variants.
Variants of uncertain significant (VUSs) are a major challenge in interpreting the results of clinical genetic testing. Because the possible effects of a VUS are unknown, the presence of a VUS should not affect clinical decision making and testing should not be offered to unaffected family members for risk assessment. Reasons for this include that, on reclassification, most VUSs will be downgraded to likely benign or benign rather than upgraded to likely pathogenic or pathogenic. In addition, because of the degree of genetic relationship, the prior probability of testing a close relative for a VUS can be as high as 50%, whereas, its presence or absence has virtually no predictive value for developing the disease in an unaffected relative. In the worst-case scenario, testing for a VUS can be misleading and lead to unnecessary treatment in relatives.17 Testing laboratories, consortia such as ClinGen (Clinical Genome Resource, https://clinicalgenome.org) and expert panels provide periodic updates to reclassify these variants to either pathogenic or benign as more data become available. This includes continued clinical testing in these individuals to provide additional phenotyping information. Patients with a VUS result should be counselled on the meaning of a VUS and anticipated reclassification. VUSs should be periodically reviewed to determine if additional evidence is available to reassign the variant.
An important caveat to the interpretation of genetic testing is that the absence of a PV does not mean a genetic aetiology does not exist. Many clinical genetic testing laboratories offer gene panels based on disease phenotype (such as an aortopathy panel or a cardiomyopathy panel), which often includes only the most common PVs known to be associated with that disease. Patients might have disease caused by a known PV that is not included in commercially available panels or might have a PV that not yet been identified. Databases of disease, such as ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), OMIM (http://www.ncbi.nlm.nih.gov/omim), the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and DECIPHER (https://decipher.sanger.ac.uk/), can be useful resources in these cases. 12
As genetic testing becomes more common in clinical practice, many companies are emerging providing direct-to-consumer genetic testing such as 23andme (www.23andme.com) or Ancestry DNA (https://www.ancestry.com/dna/). While many of these companies promise to provide insights into an individual’s risk of disease and family background, the gene panels in direct-to-consumer products are not the same as those performed by a clinical laboratory and are not a substitute for disease-specific clinical genetic testing. For example, direct to consumer might list screening for cardiomyopathy but test only for a few PVs in MYBPC3 and MYH7, rather than a more complete panel. This might lead patients to think they have had appropriate genetic testing when they have not. Concerns also have been raised about the accuracy of some of these products and government oversight and regulation are in flux. Additionally, there are significant limitations on what the direct-to-consumer testing companies are allowed to disclose about abnormalities identified during genetic testing beyond the limited panels tested.
Pathogenic variants: how does this information change patient management?
The primary reason to perform genetic testing is that knowledge of specific gene variants may change patient management, including the need and timing of imaging surveillance, the optimal choice of medical therapy, lifestyle recommendations regarding exercise,18 the risk of pregnancy and the need for surgical invention such as placement of an implantable cardiac defibrillator (ICD), aortic root surgery or orthotopic heart transplantation.19 Patients also may choose to incorporate genetic risk information into family planning. Preimplantation genetic diagnosis and prenatal genetic testing may be used to identify which pregnancies are at risk for a known familial PV.
Conversely, genetic testing showing the absence of a PV in unaffected family members avoids unnecessary medical care and provides reassurance to that family member and their offspring. Although there always is the possibility of a genetic cause that is not yet known, the absence of a known PV in a patient with a specific cardiovascular phenotype informs prognosis and may impact treatment decisions.
Aortopathies
Genetic testing results, even when negative for genes known to be associated with aortic aneurysm and dissection, allow individualised management in affected patients and in family members. (Table 1)For example, aortic imaging is recommended more frequently for patients with aortic dilation due to a PV compared with patients with hypertensive or atherosclerotic disease. The extent of vascular imaging also is affected with imaging focused only on the aortic root and ascending aorta (non-genetic and bicuspid valve disease), the entire aorta (Marfan syndrome and genetic familial aortic aneurysm) or the aorta and cerebrovascular bed (Loeys-Dietz syndrome). Imaging typically is recommended in family members who have a PV even when phenotypic features are not present because aortic dilation may be present and progressive in the absence of symptoms. In addition, the timing of preventative aortic intervention depends on the specific genetic diagnosis with current recommendations for surgery when aortic diameter reaches 45 mm in Loeys-Dietz patients, 50 mm in Marfan patients and 55 mm in bicuspid valve patients.20 21 Finally, the type of aortic surgery is impacted with replacement of the aortic sinuses (with coronary reimplantation) and stabilisation of the aortic annulus recommended for patients with a genetic aortopathy, whereas a simpler procedure with replacement of the aortic valve and ascending aorta, but preservation of the aortic sinuses and coronary artery ostia, often is reasonable when aortic dilation is not due to a single gene PV.22
Pathogenic variants strongly associated with aortic aneurysm and dissection*
Cardiomyopathies
Cardiomyopathies often are due to genetic PVs including patients with HCM, dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy (ARVC), left ventricular non-compaction and restrictive cardiomyopathy.(table 2) HCM is the most prevalent (1:500) genetic cardiomyopathy and is caused by variants in genes encoding components of the sarcomere, such as myosin heavy chain 7 (MYH7) and cardiac myosin-binding protein C (MYBPC3).23–25 Dilated cardiomyopathy is more genetically heterogeneous than HCM, with genes encoding sarcomere, cytoskeletal, nuclear lamina, calcium handling and muscular dystrophy proteins contributing to the cardiomyopathy. Testing for a core panel of DCM-causing genes is recommended and often genes causing HCM and ARVC are included due to phenotype overlap. As a result, genetic testing is guided by the additional clinical phenotypes seen with non-compaction. Genetic mutations are identified in approximately 60% of patients with ARVC, while the genetic variants found in restrictive cardiomyopathy have overlap with the genes tested for dilated cardiomyopathy. Left ventricular non-compaction is typically seen with other findings of cardiomyopathy. As a result, the appropriate genetic testing panel is guided by the additional clinical phenotypes seen with non-compaction. Restrictive cardiomyopathy is a rare form of cardiomyopathies and genetic causes of disease continue to be identified. PVs in this population are known to be in genes that cause HCM or dilated cardiomyopathy. Currently, genetic testing for cardiomyopathy facilitates family screening and identifies family members at risk for developing cardiomyopathy. There are no specific guideline recommendations for enacting a specific medical therapy or device therapy based on the results of genetic testing.26 27 However, patients with ARVC are known to have a higher prevalence of ventricular arrhythmias and heart failure with exercise. Identification of a PV in a gene known to cause ARVC would result in recommendations to restrict participation in competitive sports and high-intensity exercise.12 28 Additionally, certain genetic mutations are associated with arrhythmia, such as LMNA, and an implantable cardioverter-defibrillation can be considered before the left ventricular ejection fraction is below 35%.29
Pathogenic single-gene variants causing cardiomyopathies
Arrhythmias
In patients with cardiac arrhythmias, a combination of clinical and genetic testing can inform personalised recommendations regarding lifestyle, medications, ICD implantation or left cardiac sympathetic denervation.(table 3) (figure 4). The Heart Rhythm Society, European Heart Rhythm Association, Asia Pacific Heart Rhythm Society Expert Consensus Statement recommend that Long-QT Syndrome (LQTS) can be diagnosed based on: (1) an LQTS risk score ≥3.5, or (2) an unequivocally PV in an LQTS gene, or (3) QTc ≥500 ms on ECG in the absence of a secondary cause.30 Patients with LQTS1–3, caused respectively by a PV in the KCNQ1, KCNH2 and SCN5A genes and QTc >500 ms, are at the highest risk for lethal arrhythmias, and international LQT registries have demonstrated that ß-blocker therapy reduces lethal events by 74% in LQTS1 and by 63% in LQTS2, with less clear benefit in LQTS3.31 Known triggers that should be avoided in patients with a arrhythmia due to a PV include: QT-prolonging drugs in all individuals with LQTS, sudden loud noises (alarm clock) in LQT2 patients, strenuous exercise in catecholaminergic polymorphic ventricular tachycardia (CPVT) patients and fever in patients with Brugada syndrome.30 The first line of medical therapy for confirmed LQTS or CPVT is a ß-blocker. Decisions regarding ICD implantation depends on clinical features such as history of cardiac arrest or recurrent syncope on ß-blocker therapy. Left cardiac sympathetic denervation is effective in high-risk patients in whom ICD is relatively contraindicated, such as infants and children with high-risk LQTS.32
Pathogenic single-gene variants causing primary arrhythmias

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Example of how genetic testing might impact prevention and treatment of cardiac arrhythmias. Current model: patients may be seen by a variety of specialists for non-specific complaints such as dizziness, palpitations or syncope. Cardiac arrest may be the first contact with a cardiologist. Future model: patients at increased risk can be identified by genetic testing and family history. Cardiac imaging and electrophysiology in this subset could identify early disease. The combination of genetic test information and cardiac phenotyping can stratify patients at high, intermediate and low risk for personalised recommendations. Patients at high risk include those with: history of syncope, QTc ≥500 ms and family history of sudden cardiac death. AED, automated external defibrillator; EP, electrophysiologic; H&P, history and physical examination.
Summary
The increasing recognition that many cardiovascular diseases are due to single-gene PVs mandates that clinicians consider the possibility of an inherited cardiovascular condition when there is a positive family history or suggestive phenotypic features. Genetic diagnosis and phenotypic evaluation go hand in hand in ensuring patients receive an accurate diagnosis and optimal treatment. Cardiogenetics is a rapidly evolving field, and it is likely to contribute to improved risk stratification and management of many cardiovascular disease in the future.
Glossary of common terms
ARVC = Arrhythmogenic right ventricular cardiomyopathy
CPVT = Catecholaminergic polymorphic ventricular tachycardia
DCM = Dilated cardiomyopathy
DNA=deoxyribonucleic acid
FAA = Familial aortic aneurysm
GWAS = Genome-wide association study
HCM = Hypertrophic Cardiomyopathy
ICD = Implantable cardiac defibrillator
LMNA = Lamin A/C
LQTS = Long QT Syndrome
Lp(a) = Lipoprotein (a)
MYBPC3 = Cardiac myosin-binding protein C
MYH7 = Myosin heavy chain 7
PV = Pathogenic variant
SNP = Single nucleotide polymorphism
VUS = Variant of uncertain significance
Key points
Clinical genetic testing now is accessible for several types of inherited cardiovascular conditions.
Single nucleotide polymorphisms (SNPs) are non-pathogenic variants within the DNA sequence, with a population frequency of at least 1%.
SNPs can be used to identify pathogenetic variants in genome-wide association studies and to study causal disease mechanisms in Mendelian randomisation studies.
Pathogenic variants are nucleotide changes that result in altered production or function of the protein encoded by that gene, resulting in clinical manifestations.
Clinical and imaging evaluation for phenotypic features and a detailed family history are the initial steps in evaluation of patients who may have a cardiovascular condition due to a genetic cause.
A core tenet of clinical genetics is to begin genetic testing in a clearly affected patient (the index case or proband) to maximise the likelihood of identifying a pathologic variant.
If a pathological variant has been identified, genetic testing of other family members should be limited to the single pathogenic gene.
Common reasons for genetic testing in clinical cardiology are phenotypic features, a clinical event or a family history suggestive of an inherited condition in patients with aortopathy, cardiomyopathy or an arrhythmia.
Identification of a pathogenic variant often changes clinical management in terms of need and timing of imaging surveillance, optimal choice of medical therapy lifestyle and exercise recommendations, family planning and risk of pregnancy, and timing and type of surgical intervention.
Identification of a variant of uncertain significance should not affect clinical decision making, and testing should not be offered to unaffected family members for risk assessment.
Direct-to-consumer genetic testing might provide insights into family background and risk of disease in general but does not provide screening for pathogenic variants causing specific cardiovascular conditions.
CME credits for Education in Heart
Education in Heart articles are accredited for CME by various providers. To answer the accompanying multiple choice questions (MCQs) and obtain your credits, click on the ‘Take the Test’ link on the online version of the article. The MCQs are hosted on BMJ Learning. All users must complete a one-time registration on BMJ Learning and subsequently log in on every visit using their username and password to access modules and their CME record. Accreditation is only valid for 2 years from the date of publication. Printable CME certificates are available to users that achieve the minimum pass mark.
References
Footnotes
Twitter @jainysavlamd
Contributors All of the authors drafted and edited the manuscript, and all authors approved the final version.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Author note References which include a * are considered to be key references