Article Text

Statistics from Altmetric.com
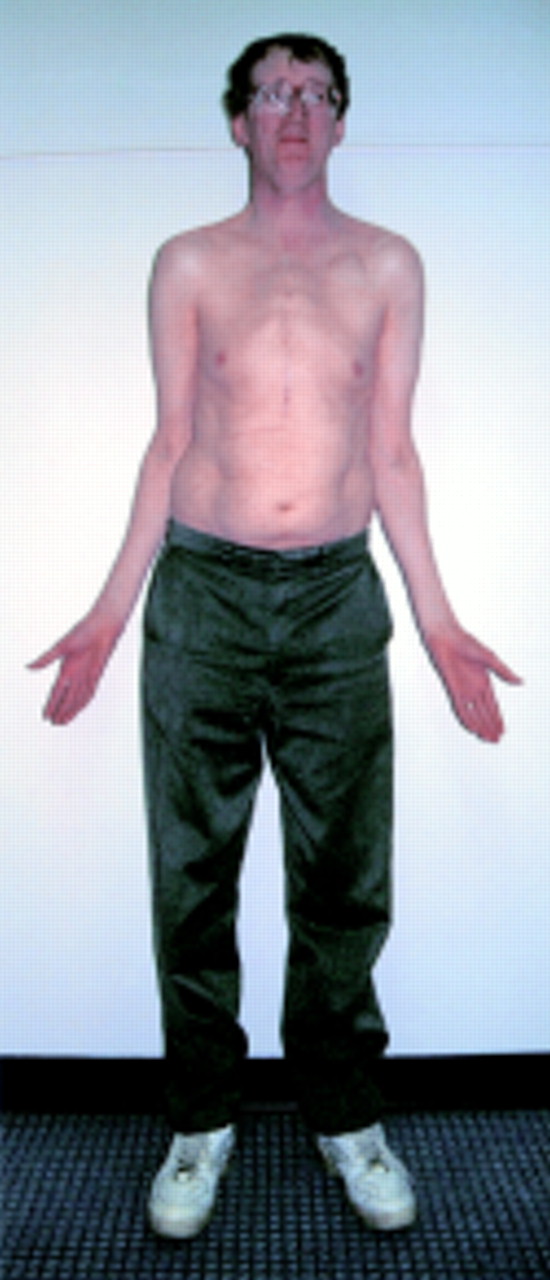
Marfan syndrome is a variable, autosomal dominant connective tissue disorder, affecting mainly the cardiovascular system, eyes, and skeleton. The incidence is approximately 1 in 9800, and around 26% of cases have no family history, the condition resulting from a new mutation.1 Characteristic features include progressive aortic dilatation associated with aortic valve incompetence, mitral valve prolapse and incompetence, lens dislocation and myopia, and a tall and thin body (fig 1) with long limbs, arachnodactyly, pectus deformities, and sometimes scoliosis. Further less specific features are often detectable in the clinic, such as a high palate with dental crowding, and skin striae distensae, and other characteristic findings may be sought by radiological imaging, such as protrusio acetabulae and dural ectasia. A history of recurrent pneumothorax may be found in some cases. The clinical features have been codified into the so-called Ghent diagnostic nosology,2 as the clinical variability of the condition can otherwise make diagnosis difficult.

Patient with Marfan syndrome. Note aesthenic or marfanoid body habitus, pronounced myopia (thick glasses), previous thoracic surgery (composite aortic root/aortic valve replacement). Residual evidence of pectus carinatum can be seen. Photograph reproduced with patient's permission.
Life expectancy is primarily determined by the severity of cardiovascular involvement, and has improved substantially in the past 30 years as a result of improved medical and surgical management. In particular, β blockade reduces the rate of aortic dilatation in some patients, and, perhaps not surprisingly, the outcome of prophylactic aortic root surgery has been shown in several recent series to be superior to that of emergency surgery for dissecting aneurysm. The timing of prophylactic surgical intervention depends on a number of factors including the aortic diameter and its rate of dilatation, implying a need for regular aortic root surveillance. The optimal management of Marfan patients may therefore require lifelong medical treatment, and lifelong aortic surveillance with a view to potential aortic root surgery, representing a major commitment for patient and doctor alike. At the same time, a diagnosis of Marfan syndrome may have serious social consequences for the patient, in terms of lifestyle, employment, and insurance.
Marfan syndrome almost always results from mutation in the fibrillin 1 gene on chromosome 15,3 although in one family the disease was linked to an unknown gene on chromosome 3. Molecular testing for Marfan syndrome has proved less useful than was hoped for two main reasons. Firstly, very few fibrillin 1 mutations have been observed more than once, so the detection of a mutation yields little prognostic information beyond that available from the patient's own family history. Secondly, fibrillin 1 mutations have also been detected in Marfan related disorders whose cardiovascular involvement is milder or non-existent. These include MASS syndrome, a disorder with some Marfan-like features (myopia, mitral valve prolapse, aortic dilatation, skin involvement, skeletal involvement) but mild and apparently stable aortic dilatation, and isolated ectopia lentis.4 The importance of a careful clinical assessment and accurate clinical diagnosis cannot therefore be overstated, if appropriate targeting of medical and surgical resources in Marfan patients is to be achieved. In this article, I will review current approaches to diagnosis of Marfan syndrome and cardiovascular management.
▸ MAKING THE DIAGNOSIS OF MARFAN SYNDROME
Much effort has been expended over the years in devising agreed diagnostic criteria for Marfan syndrome. The currently accepted criteria, known as the Ghent nosology,2 were defined in 1996 as a revision of the earlier Berlin criteria of 1988. In the Ghent nosology, clinical features in the skeletal, ocular, cardiovascular, pulmonary, and integumentary systems are used to define either a major criterion or only involvement of a particular organ system (table 1). The dura and the genetic findings count as two further systems, with lumbosacral dural ectasia on imaging studies, and aspects of the family history or genetic testing being classified as a major criterion in each respectively. A diagnosis of Marfan syndrome requires, as a minimum, a major criterion in two systems and involvement of a third.
Ghent diagnostic nosology
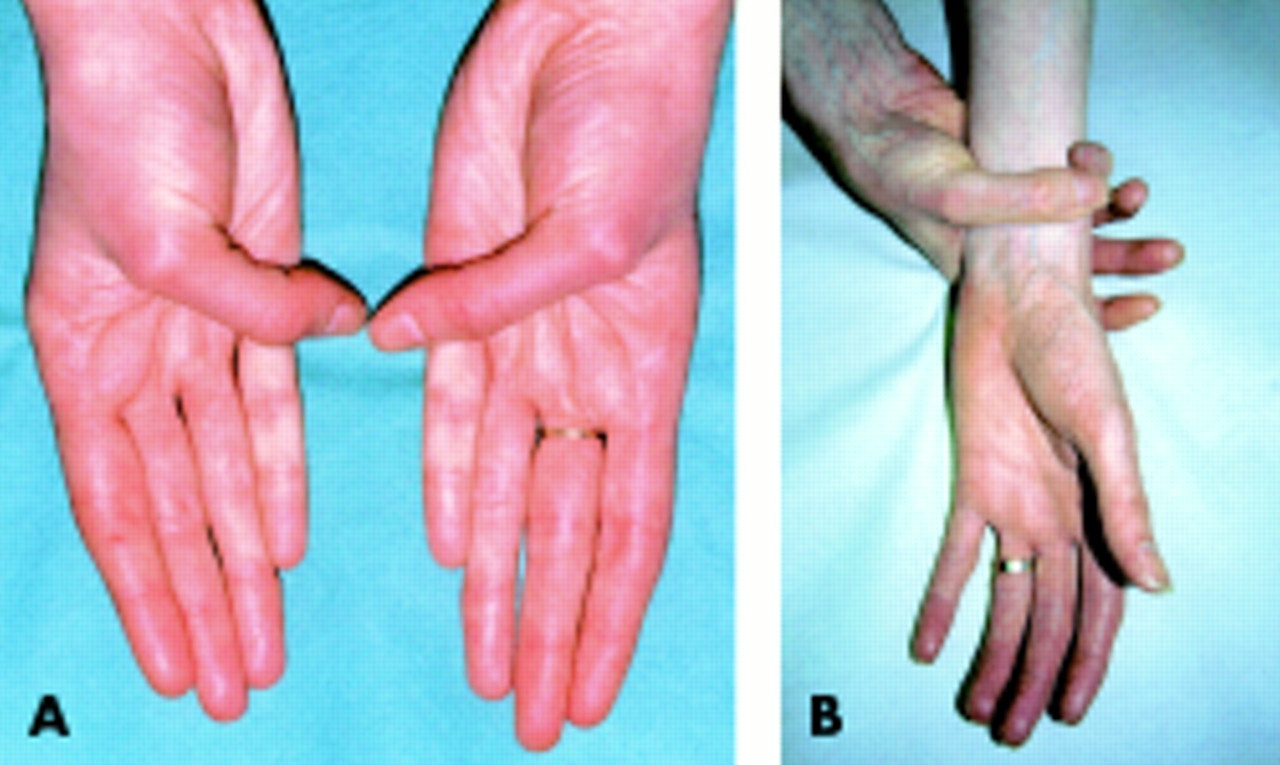
In the skeletal system, arachnodactyly may be assessed (fig 2) using the Steinberg thumb sign (the entire thumbnail projects beyond the ulnar border of the hand) and the Walker-Murdoch wrist sign (the thumb and fifth finger overlap around the wrist). An upper to lower segment ratio in an adult of less than 0.86, or a span to height ratio of greater than 1.05, are objective measures of a marfanoid body habitus. The lower segment is measured as the distance from the symphysis pubis to the floor, and the upper segment calculated by subtracting this distance from the height. One of the eight major skeletal features in the Ghent nosology (four must be present for the skeletal system to contribute a major criterion) is protrusio acetabulae, detected by pelvic x ray. To avoid unnecessary x ray exposure, this investigation should only be undertaken when a positive finding would make the diagnosis of Marfan syndrome under the Ghent nosology. Lumbosacral magnetic resonance imaging (MRI) scanning for dural ectasia should be considered in similar circumstances.

Arachnodactyly. (A) Steinberg thumb sign. Entire thumb nail protrudes beyond ulnar border of hand. (B) Walker-Murdoch wrist sign. Thumb and fifth finger can overlap around wrist. Both signs must be present to diagnose arachnodactyly according to the Ghent Marfan diagnostic criteria.
In the ocular system, only lens dislocation is regarded as a major criterion, other less specific findings such as myopia being regarded as eye involvement.
Aortic dilatation or dissection are the major criteria in the cardiovascular system. Aortic diameter should be measured by transthoracic echocardiogram at the sinuses of Valsalva (fig 3) and related to normal values based on age and body surface area (BSA).2,5 The risk of aortic dissection rises as the aortic root diameter increases.6 Other imaging techniques such as transoesophageal echocardiography, MRI or computed tomographic scanning (fig 4) may be helpful in some cases.

Diagram of the aortic root as seen at echocardiography. The aortic diameter may be measured at the aortic annulus (1), the sinuses of Valsalva (2), the supra-aortic ridge (3), and the proximal ascending aorta (4). In Marfan syndrome, dilatation usually starts at the sinuses of Valsalva, so this measurement is critical in monitoring the early evolution of the condition. Diameters must be related to normal values for age and body surface area. After Roman et al, Ann Intern Med 1987;106:800–7, with permission.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Parasagittal breath hold T1 magnetic resonance (MR) image showing pronounced dilatation of the aortic root with slight dilatation of the descending aorta in a young adult with Marfan syndrome. Line 1 represents the diameter at the sinuses of Valsalva. (B) Parasagittal cine MR angiogram showing dilated aortic root but with normal upper ascending arch and descending aorta in a young adult. (C) Axial computed tomographic (CT) scan at T7 of a Marfan patient showing dilated ascending and descending aorta with dissection flap anteriorly in the descending aorta and previous surgery to the ascending aorta. (D) Parasagittal reformatted CT of chest and abdomen in the same patient with contrast showing dilatation of the whole of the aorta with a spiral dissection from the arch through to the lower abdominal aorta. MR and CT images courtesy of Professor J Weir, Department of Radiology, Grampian University Hospitals NHS Trust.
Many Marfan features such as echocardiographic findings,7 ectopia lentis, scoliosis, upper to lower segment ratio, and protrusio acetabulae are age dependent in their occurrence.8 For younger patients with a family history of Marfan syndrome who do not fulfil the diagnostic criteria, and for younger Marfan-like patients with no family history who only fail to meet the diagnostic criteria by one system, repeat evaluations should be considered until age 18 (see box). This is to avoid missing the evolving diagnosis, while not stigmatising children and adolescents who may be unaffected. From the foregoing discussion, it can be seen that initial evaluation of patients with possible Marfan syndrome requires a multidisciplinary approach involving clinical genetics, cardiology, ophthalmology, and radiology.
DIFFERENTIAL DIAGNOSIS
There are a number of disorders with similar features to Marfan syndrome, but some can be easily distinguished by their other clinical features or by biochemical testing. Lujan-Fryns syndrome is an unusual X-linked mental handicap disorder with marfanoid features, while in the autosomal dominant Shprintzen-Goldberg syndrome, craniosynostosis is also evident. Some Shprintzen-Goldberg cases have fibrillin 1 mutations, so this might be regarded as an unusual variant of Marfan syndrome. Homocystinuria (autosomal recessive) is characterised by raised urinary homocysteine excretion, while in congenital contractural arachnodactyly or Beals syndrome (autosomal dominant, associated with mutation in fibrillin 2) joint contractures and ear anomalies are evident in addition to a marfanoid appearance. In other disorders, the distinction from Marfan syndrome is more difficult. The best known of these is the MASS syndrome, which may be considered a forme fruste of Marfan syndrome. The name is an acronym for the clinical features (myopia, mitral valve prolapse, aortic dilatation, skin involvement, skeletal involvement), the skin and skeletal features being similar to those seen in Marfan syndrome, but less pronounced. The disorder may be associated with fibrillin-1 mutation,3,4 but the aortic dilatation is mild, and the risk of aortic aneurysm rupture seems low.
Assessment of a patient with possible Marfan syndrome
-
The diagnosis should be based on the Ghent diagnostic nosology
-
The initial assessment should include a personal history, detailed family history, and clinical examination including ophthalmology examination and transthoracic echocardiogram
-
The aortic diameter at the sinus of Valsalva (fig 3) should be related to normal values based on age and body surface area
-
The development of scoliosis and protrusio acetabulae is age dependent, commonly occurring following periods of rapid growth. Radiographic examination for these features is indicated, depending on age, if a positive finding would make the diagnosis of Marfan syndrome
-
A pelvic MRI scan to detect dural ectasia is indicated if a positive finding would make the diagnosis of Marfan syndrome
-
Younger patients with suspected Marfan syndrome, who do not fulfil the Ghent diagnostic criteria, should be offered repeat clinical evaluations pre-school, before puberty, and at age 18, if they fall into one of the groups below. Additional evaluations may be clinically indicated around puberty:
Children or adolescents with a positive family history in whom DNA testing is not possible
Children or adolescents with no family history, who fall short of fulfilling the diagnostic criteria by one system only.
A condition of mitral valve prolapse with thoracic skeletal anomalies (often pectus excavatum) but without other Marfan features has also been described, and in one case a fibrillin 1 mutation was found.4 Molecular investigation of this disorder has not been widely undertaken, and it is not clear whether it is truly distinct from MASS syndrome. Autosomal dominant ectopia lentis may occur with Marfan-like skeletal findings, fibrillin 1 mutation, but no cardiac involvement. There are also forms of hereditary aortic aneurysm without other Marfan features, which have been associated with as yet unidentified genes on chromosomes 5 and 11. Other hereditary connective tissue disorders, such as Ehlers-Danlos syndrome and Stickler syndrome, may also cause confusion because of their overlapping features. Skin laxity and easy bruising are prominent in the Ehlers-Danlos syndrome while myopia with retinal detachment but not ectopia lentis is a feature of Stickler syndrome. Stickler syndrome patients also have a distinctive facial appearance (midface hypoplasia), and the condition is sometimes associated with cleft palate and hearing loss.
CARDIOVASCULAR COMPLICATIONS IN MARFAN SYNDROME
Cardiovascular complications of Marfan syndrome include mitral valve prolapse and regurgitation, left ventricular dilatation and cardiac failure, and pulmonary artery dilatation, but aortic root dilatation is the most common cause of morbidity and mortality. Aortic valve incompetence usually arises in the context of a dilated aortic root, and the risk of aortic rupture increases substantially when the diameter at the sinus of Valsalva exceeds 5.5 cm.5,9 Myocardial infarction may occur if an aortic root dissection occludes the coronary ostia. Marfan syndrome mortality from complications of aortic root dilatation has decreased (70% in 1972, 48% in 1995) and life expectancy has increased (mean (SD) age at death 32 (16) years in 1972 versus 45 (17) years in 1998),1 associated with increased medical and surgical intervention. Risk factors for aortic dissection include increased aortic diameter, extent of aortic dilatation, rate of aortic dilatation, and family history of aortic dissection.7,9
LABORATORY FINDINGS
Histologically, the Marfan aorta is characterised by elastic fibre fragmentation and disarray, paucity of smooth muscle cells, and deposition of collagen and mucopolysaccharide between the cells of the media. These appearances are sometimes described as “cystic medial degeneration” although there are no true cysts present. Mucopolysaccharide deposition in the valves may cause valve leaflet thickening. The molecular basis for these changes is abnormality of fibrillin, an important component of the elastic microfibril. Fibrillin is a 350 kD glycoprotein, synthesised as a 375 kD precursor which is processed and secreted into the matrix. It is encoded by the fibrillin 1 gene which maps to chromosome 15q21.1. Mutation in another fibrillin gene (fibrillin 2, mapping to chromosome 5q23–31) causes Beals syndrome. With current techniques, fibrillin 1 mutations can be detected in about 66% of Marfan patients, diagnosed by the Ghent nosology.4 Most mutations are missense, suggesting that the phenotype is usually cause by a dominant negative effect of the mutant gene product on microfibrillar assembly. Some nonsense and frameshift mutations are also seen, suggesting that reduced amount of fibrillin (haploinsufficiency) can also cause Marfan syndrome. Wide variability in severity has been documented with different mutations, even in the same codon, causing either severe neonatal Marfan syndrome or classical adult Marfan syndrome. Similarly, mutations in the central region of the gene (exons 24–32), sometimes called the “neonatal region”, may be associated with phenotypes ranging from severe neonatal Marfan syndrome to isolated ectopia lentis.
PATHOPHYSIOLOGY AND MEDICAL MANAGEMENT
The pathophysiological consequence of the elastic fibre degeneration seen in the Marfan aorta is reduced distensibility in response to the pulse pressure wave or increased stiffness, and this can be detected at any age by echocardiography10 or gated MRI scanning, although it is less pronounced in children. This abnormal aortic compliance is associated with an increased pulse wave velocity. The normal aorta dilates gradually with age, and becomes stiffer, but these changes are more notable at any age in Marfan syndrome. In the early 1970s, it was suggested that reduction of the systolic ejection impulse (dP/dt, the rate of change of central arterial pressure with time) using β blocker treatment might reduce the risk of aortic dissection in Marfan syndrome.7 Studies in turkeys prone to aortic dissection had shown improved survival following treatment with propranolol. A randomised trial of propranolol treatment in 70 adolescent and young adult Marfan patients demonstrated a reduced rate of aortic dilatation and fewer aortic complications in the treatment group, although some patients seemed to respond better than others.7 A retrospective historically controlled trial of propranolol or atenolol treatment in 113 patients found similar effects.11 Recent studies have shown that β blockade with propranolol, atenolol or metoprolol increases aortic distensibility, and reduces aortic stiffness and pulse wave velocity9,12,13 in a subgroup of Marfan patients, although in the “non-responders” a minor deterioration in aortic distensibility was seen, the clinical significance of which is unclear. Those who responded to β blockade tended to have smaller aortic diameters12 (< 4 cm in one study10), in keeping with other studies suggesting that the reduction in rate of aortic dilatation with β blockade is greatest in younger patients with smaller aortas.7,11 These studies provide strong evidence that β blockade should be considered in all Marfan patients, but particularly in the younger age group (see box).
Although remarkably few withdrawals because of side effects were recorded in these studies, β blockers are not suitable for everyone—for example, patients with asthma, cardiac failure or bradyarrhythmias. Other treatments aimed at reducing the ejection impulse, such as calcium antagonists or angiotensin converting enzyme (ACE) inhibitors, have been suggested for such cases. Unfortunately, there are no reported clinical trials to confirm the benefit or otherwise of these drugs in Marfan syndrome. There may be theoretical reasons to consider ACE inhibitors or angiotensin II receptor blockers for future clinical study. Vascular smooth muscle cell apoptosis has been implicated in the cystic medial degeneration seen in the Marfan aorta, and both types of drug have been shown to inhibit vascular smooth muscle cell apoptosis in cultured Marfan aortic media cells.14 In addition, one small study demonstrated abnormal flow mediated vasodilation of the brachial artery in Marfan patients, although agonist mediated vasodilation was normal. This was attributed to abnormal endothelial cell mechanotransduction associated with abnormal fibrillin. This could lead to alternative molecular targets for future pharmacological interventions. There is much scope for further laboratory and clinical trial work in this area.
Cardiovascular management of Marfan syndrome
-
β Blocker treatment should be considered in any Marfan patient with aortic dilatation at any age, but prophylactic treatment may be more effective in those with an aortic diameter of < 4 cm
-
Risk factors for aortic dissection in Marfan syndrome include aortic diameter > 5 cm, aortic dilatation extending beyond the sinus of Valsalva, rapid rate of aortic dilatation (> 5% per year, or 2 mm/year in adults), and family history of aortic dissection
-
Marfan patients of all ages should be offered at least annual evaluation with clinical history, examination, and transthoracic echocardiography. In children, serial transthoracic echocardiography at 6–12 month intervals is recommended, the frequency depending on the aortic diameter (in relation to body surface area) and the rate of increase
-
Marfan patients should be referred for prophylactic aortic root surgery when the diameter at the sinus of Valsalva exceeds 5.5 cm in an adult or 5.0 cm in a child
-
Pregnant patients with Marfan syndrome are at increased risk of aortic dissection if the aortic diameter exceeds 4 cm. Such cases warrant frequent cardiovascular monitoring throughout pregnancy and into the puerperium
SURGICAL MANAGEMENT
Aortic root dilatation leading to aortic dissection and/or aortic valve dysfunction usually occurs first at the sinus of Valsalva.5 There is general agreement, based on a number of comparative studies, that there is a better outcome with early aortic root surgery than with later or emergency surgery,9,15 and prophylactic surgery is recommended when the diameter at the sinus of Valsalva exceeds 5.5 cm in an adult2,9 and 5.0 cm in a child. Other factors such as the rate of growth of the aortic diameter and family history of aortic dissection may be taken into account, and it may be that assessment of aortic distensibility will become a useful prognostic indicator. Survival data also show improved longevity of Marfan patients who undergo prophylactic surgery, compared with their untreated relatives.16 Not surprisingly, part of this is attributable to death before reaching hospital in untreated patients with acute dissection. Until recently, the procedure of choice for most cardiothoracic surgeons would have been the Bentall composite graft repair, in which both the aortic root and the aortic valve are replaced. This procedure has a low operative mortality in experienced hands, with long term survival of around 80% at five years and 60% at 10 years.17 In 1979, Yacoub and colleagues described a valve conserving (remodelling) technique which therefore avoids the need for long term anticoagulants, and in 1992 David and Feindell described an alternative valve conserving procedure in which the native aortic valve is re-implanted in a Dacron tube (re-implantation technique).
Application of these methods in Marfan syndrome has been controversial, as it has been suggested that further deterioration of the aortic valve leaflets will inevitably require reoperation for valve replacement at a later date. Case series currently being reported suggest that when use of these procedures is restricted to cases where the aortic valve appears structurally normal and valve incompetence is largely caused by annular ectasia, the long term outcome is as good as the Bentall procedure, without the hazards of anticoagulation.18 There is certainly a case for considering this option for children (where future growth is also a consideration), women, and in those in whom anticoagulation may be hazardous. As Marfan patients survive longer, reoperation for new aneurysms developing elsewhere in the arterial tree are becoming common (for example, see fig 4C,D)—in one series, 70% developed second aneurysms requiring surgery.16 Partly for this reason, continuation of long term β blockade after surgery is strongly recommended in most Marfan centres.17 Marfan syndrome may also affect the function of other heart valves and give rise to symptoms for which valve surgery may be indicated in children and adults.2 For example, mitral valve replacement may be required in up to 10% of those requiring aortic root surgery.16
CARDIOVASCULAR FOLLOW UP
It is clear that with the need for life long β blocker treatment, the possible development of other medical therapies for those who cannot tolerate β blockers, and the advantages of prophylactic aortic root surgery based on the size and rate of change of the aortic diameter, lifelong follow up is advisable for Marfan patients. At least annual evaluation with clinical history and examination and transthoracic echocardiography5 is recommended, with additional assessments as clinically indicated (see box). The rate of change of the aortic diameter should clearly influence follow up intervals.
MARFAN SYNDROME AND PREGNANCY
The two major issues in pregnancy are the risk of cardiovascular complications in an affected mother and the 50% risk of transmission of Marfan syndrome to the fetus. The risk of aortic dissection in pregnancy is increased, and may be caused by inhibition of collagen and elastin deposition in the aorta by oestrogen, and the hyperdynamic hypervolaemic circulatory state of pregnancy. Gestational hypertension and pre-eclampsia may increase the risk of aortic rupture. Most complications occur in women with pre-existing cardiac disease. In three recent studies, two retrospective and one prospective, nine women in 83 (11%) had severe complications, mostly aortic rupture, although endocarditis was also reported. Cardiovascular complications appear more likely if the aortic root is greater than 4 cm at the start of pregnancy, or dilates rapidly.19 β Blockers should be continued throughout pregnancy. With regard to the risk of transmission of Marfan syndrome to the fetus, genetic counselling should be offered to women of childbearing age with Marfan syndrome. Mutation detection or linkage can be used for prenatal diagnosis in some families if the parents wish, but fetal ultrasound scanning is unreliable.
MARFAN SYNDROME AND SPORTS
The risks of sports in patients with Marfan syndrome depend on which organ systems are involved and to what degree, in a particular patient. Low to moderate activity levels have been regarded as acceptable for Marfan patients. Contact sports involving a likelihood of bodily collision (for example rugby, high diving, equestrian events) may not be advisable, both because of cardiovascular risks and because of the risk of exacerbating lens dislocation. Patients who have undergone aortic root or valve replacement, or both, will have more severe restrictions, particularly if taking anticoagulants. Scuba diving carries a risk of precipitating pneumothorax in Marfan patients and should be avoided.
CONCLUSION
The investigation, management, and long term follow up of patients with Marfan syndrome is complex and requires a multidisciplinary approach. Although many studies have been carried out into the delineation and management of Marfan syndrome, the trials and case series generally involve relatively few patients, the largest being in the hundreds. For those used to evidence based clinical management based on trials of thousands, this may present a difficulty, which has been recognised in a recent discussion of evidence based management of genetic conditions.20 Despite this, the outlook for Marfan patients has improved dramatically since the 1970s, and this should inspire further endeavours for Marfan patients as there are many aetiological and management issues still to be resolved.
Acknowledgments
This article is based on work undertaken in conjunction with colleagues from many disciplines who formed the Marfan syndrome Guideline Development Group (see below) as part of a project funded by the Clinical Resources and Audit Group of the Scottish Executive Department of Health.
Membership of the Marfan Syndrome Guideline Development Group
Clinical genetics: JCS Dean, NE Haites, E Hobson, Z Miedzybrodzka, S Moore, S Simpson. Anaesthetics: P Martin. Cardiology: P Booth (paediatrics), S Walton (adults). Cardiothoracic surgery: JS Cockburn, R Jeffrey. Obstetrics: M Hall. Ophthalmology: W Church. Orthopaedics: T Scotland. Respiratory medicine: JAR Friend (adults), G Russell (paediatrics), Rheumatology: D Reid. Scottish Guideline Steering Group: N Bradshaw, H Campbell, M Porteous.
REFERENCES
Supplementary materials
Web-only References1. Murdoch JL, Walker BA, Halpern BL, Kuzma JW, McKusick VA. Life expectancy and causes of death in the Marfan syndrome. N Engl J Med 1972; 286:804-808
2. Silverman DI, Burton KJ, Gray J, Bosner MS, Kouchoukos NT, Roman MJ, Boxer, M, Devereux RB, Tsipouras P. Life expectancy in the Marfan syndrome. Am J Cardiol 1995;75(2):157-60
3. Collod G, Babron MC, Jondeau G, Coulon M, Weissenbach J, Dubourg O, Bourdarias JP, Bonaiti Pellie C, Junien C, Boileau C. A second locus for Marfan syndrome maps to chromosome 3p24.2-p25. Nature Genet 1994;8:264-268
4. Beighton P, De Paepe A, Danks G, Finidori G, Gedde-Dahl T, Goodman R, Hall JG, Hollister DW, Horton W, McKusick VA et al. International nosology of heritable disorders of connective tissue, Berlin 1986. Am J Med Genet 1988; 29: 581-594
5. Silverman DI, Gray J, Roman MJ, Bridges A, Burton K, Boxer M, Devereux RB, Tsipouras P. Family history of severe cardiovascular disease in Marfan syndrome is associated with increased aortic diameter and decreased survival. JACC 1995;26:1061-1067
6. Jeremy RW, Huang H, Hwa J, McCarron H, Hughes CF, Richards JG. Relation between age, arterial distensibility, and aortic dilatation in the Marfan syndrome. American Journal of Cardiology 1994;74:369-73
7. Adams JN, Brooks M, Redpath TW, Smith FW, Dean J, Gray J, Walton S, Trent RJ. Aortic distensibility and stiffness index measured by magnetic resonance imaging in patients with Marfan�s syndrome. Br Heart J 1995;73:265-269
8. Marsalese DL, Moodie DS, Vacante M, Lytle BW, Gill CC, Sterba R, Cosgrove DM, Passalacqua M, Goormastic M, Kovacs A. Marfan�s syndrome: natural history and long-term follow-up of cardiovascular involvement. J Am Coll Cardiol 1989;14:422-8
9. Maumenee IH. The eye in the Marfan syndrome. Tr. Am. Opth. Soc. 1981; 79: 684-733
10. Pyeritz RE, McKusick VA. The Marfan syndrome: Diagnosis and management. N Engl J Med 1979;300:14, 772-777 11. De Paepe A. Dural ectasia and the diagnosis of Marfan�s syndrome. Lancet 1999;354:878-879
12. Yule SR, Hobson EE, Dean JCS, Gilbert FJ. Protrusio acetabuli in Marfan�s syndrome. Clin Radiol 1999;54:95-97
13. Pyeritz RE, Wappel MA. Mitral valve dysfunction in the Marfan syndrome. Clinical and echocardiographic study of prevalence and natural history. Am J Med 1983;74:797-807
14. Joseph KN, Kane HA, Milner RS, Steg NL, Williamson MB Jr, Bowen JR. Orthopaedic aspects of the Marfan Syndrome. Clinical Orthopaedics and Related Research 1992; 277: 251-261
15. Hwa J, Richards JG, Huang H, McKay D, Pressley L, Hughes CF, Jeremy, RW. The natural history of aortic dilatation in Marfan syndrome. Medical Journal of Australia 1993;158:558-62
16. Cohn LH, Rizzo RJ, Adams DH, Aranki SF, Couper GS, Beckel N, Collins, JJ Jr. Reduced mortality and morbidity for ascending aortic aneurysm resection regardless of cause. Annals of Thoracic Surgery 1996;62:463-8
17. Farah MG, Suneja R Diagnosis of circumferential dissection of the ascending aorta by transesophageal echocardiography. Chest 1993;103:291-2
18. Lehmann ED. Cardiovascular screening in Marfan�s syndrome. Lancet 1995;345:1501-2
19. Putnam EA, Zhang, H, Ramirez F, Milewicz DM. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nature Genetics 1995;11:456-458
20. Lacombe D, Bonneau D, Verloes A, Couet D, Koulischer L, Battin J. Lujan-Fryns syndrome (X- linked mental retardation with marfanoid habitus): report of three cases and review. Genetic Counselling 1993; 4: 193-198
21. Saal HM, Bulas DI, Allen JF, Vezina G, Walton D, Rosenbaum KN. Patient with craniostenosis and Marfanoid phenotype (Shprintzen-Goldberg Syndrome) and cloverleaf skull. Am J Med Genet 1995;57:573-578
22. Sood S , Eldadah ZA, Krause WL, McIntosh I, Dietz HC. Mutation in fibrillin-1 and the Marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nature Genetics 1996;12:209-11.
23. Stahl-Hallengren C, Ukkonen T, Kainulainen K, Kristofersson U, Saxne T, Tornqvist K, Peltonen L. An extra cysteine in one of the non-calcium-binding epidermal growth factor-like motifs of the FBN1 polypeptide is connected to a novel variant of Marfan syndrome. J Clin Invest 1994;94:709-713
24. Simpson CF, Kling JM, Palmer RF. Beta-Aminopropionitrile-induced dissecting aneurysms of turkeys: treatment with propranolol. Toxicol Appl Pharmacol 1970;16:143-153
25. Simpson CF, Boucek RJ, Noble NL. Influence of d-,l- and dl-propranolol, and practolol on beta-amino-propionitrile-induced aortic ruptures of turkeys. Toxicol Appl Pharmacol 1976;38:169-175
26. Reed CM, Fox ME, Alpert BS. Aortic biomechanical properties in paediatric patients with the Marfan syndrome, and the effects of atenolol. Am J of Cardiol 1993;71:606-8
27. Tsang VT, Pawade A , Karl TR, Mee RB. Surgical management of Marfan syndrome in children. Journal of CardiacSurgery 1994;9:50-4.
28. Habbal MH. Cardiovascular manifestations of Marfan�s syndrome in the young. Am Heart J 1992;123:752-7
29. Pyeritz RE, Francke U. The second international symposium on the Marfan syndrome. Am J Med Genet 1993; 47: 127-135
30. Hall JR, Pyeritz RE, Dudgeon DL, Haller JA. Pneumothorax in the Marfan syndrome: prevalence and therapy. AnnThoracic Surgery 1984;37:500-504
31. Wolinsky H. Effects of estrogen and progestogen treatment on the response of the aorta of male rats to hypertension. Morphological and chemical studies. Circ Res 1972; 30: 341 - 349.
32. Lipscomb KJ, Clayton Smith J, Clarke B, Donnai P, Harris R . Outcome of pregnancy in women with Marfan�s syndrome. Br J Obstet Gynaecol 1997; 104: 201 - 206.
33. Rossiter JP, Repke JT, Morales AJ, Murphy EA, Pyeritz RE. A prospective longitudinal evaluation of pregnancy in the Marfan syndrome. Am J Obstet Gynaecol 1993; 173: 1599 - 1606.
34. Korkko J, Kaitila I, Lonnqvist L, Peltonen L, Ala-Kokko L. Sensitivity of conformation gel electrophoresis in detecting mutations in Marfan syndrome and related conditions. J Med Genet 2002;39:34-41
35. Roman MJ, Devereux RB, Kramer-Fox R, Spitzer MC. Comparison of cardiovascular and skeletal features of primary mitral valve prolapse and Marfan syndrome. Am J Cardiol 1989;63:317-321
36. Kainulainen K, Karttunen L, Puhalla L, Sakai LY, Peltonen L. Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome. Nature Genet 1994;6:64-69
37. Guo D, Hasham S, Kuang S, Vaughan CJ, Boerwinkle E, Chen H, Abuelo D, Dietz HC, Basson CT, Shete SS, Milewicz DM. Familial aortic aneurysms and dissections. Genetic heterogeneity with a major locus mapping to 5q13-14. Circulation 2001;103:2461-2468
38. Vaughan CJ, Casey M, He J, Veugelers M, Henderson K, Guo D, Campagna R, Roman MJ, Milewicz DM, Devereux RB, Basson CT. Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation 2001;103:2469-2475
39. Groenink M, de Roos A, Mulder BJM, Verbeeten B, Timmermans J, Zwinderman AH, Spaan JAE, van der Wall EE. Biophysical properties of the normal-sized aorta in patients with Marfan syndrome: evaluation with MR flow mapping. Radiology 2001;219:535-540
40. Pyeritz RE. The Marfan Syndrome. In Royce PM and Steinmann BE eds, Connective tissue and its heritable disorders. New York: Wiley-Liss 1993:455-456
41. Roberts WC, Honig HS. The spectrum of cardiovascular disease in the Marfan syndrome: a clinico-morphologic study of 18 necropsy patients and comparison to 151 previously reported necropsy patients. Am Heart J 1982;104:115-135
42. Maslen CL, Glanville RW. The molecular basis of Marfan syndrome. DNA and Cell Biology 1993;12:561-572
43. Jeremy RW, Huang H, Hwa J, McCarron H, Hughes CF, Richards JG. Relation between age, arterial distensibility and aortic dilatation in the Marfan syndrome. Am J Cardiol 1994;74:369-373
44. Haouzi A, Berglund H, Pelikan PCD, Maurer G, Siegel RJ. Heterogeneous aortic response to acute beta-adrenergic blockade in Marfan syndrome. Am Heart J 1997;133:60-63
45. Wilson DG, Bellamy MF, Ramsey MW, Goodfellow J, Brownlee M, Davies S, Wilson JF, Lewis MJ, Stuart AG.
Endothelial function in Marfan syndrome. Circulation 1999;99:909-915
46. Gelsomino S, Frassani R, Porreca Land, Livi U. Aortic repair in Marfan�s syndrome: prevention of mechanical leaflets damage. Cardiovasc Surg 2001;9:299�301
47. Alexiou C, Langley SM, Charlesworth P, Haw MP, Livesey SA, Monro JL. Aortic root replacement in patients with
Marfan�s syndrome: the Southampton experience. Ann Thorac Surg 2001;72:1502� 1508
48. Adams JN, Trent RJ. Aortic complications of Marfan�s syndrome. Lancet 1998;352:1722-1723
49. Rossi-Foulkes R, Roman MJ, Rosen SE, Kramer-Fox R, Ehlers KH, O'Loughlin JE, Devis JG, Devereux RB. Phenotypic features and impact of beta blocker or calcium agonist therapy on aortic lumen size in the Marfan syndrome. Am J Cardiol 1999;83:1364-1368
Linked Articles
- Miscellanea
- Miscellanea