Article Text

Statistics from Altmetric.com
Virtually all regional acute myocardial infarcts are caused by thrombosis developing on a culprit coronary atherosclerotic plaque. The very rare exceptions to this are spontaneous coronary artery dissection, coronary arteritis, coronary emboli, coronary spasm, and compression by myocardial bridges. Thrombosis is also the major initiating factor in unstable angina, particularly when rest pain is recent and increasing in severity. Necropsy studies suggest that a new thrombotic coronary event underlies 50–70% of sudden deaths caused by ischaemic heart disease.
The culprit plaque
Given the importance of thrombosis as the trigger for acute myocardial ischaemia, it is necessary to know something about the structure of plaques before thrombotic events occur and why there should be a sudden change from a stable state (no thrombus) to an unstable state (thrombus).
The fully developed human fibrolipid plaque, designated by the American Heart Association (AHA) as type IV or type Va,1 has a core of lipid surrounded by a capsule of connective tissue (fig 1). The core is an extracellular mass of lipid containing cholesterol and its esters, some of which is in a crystalline form. The core is surrounded by numerous macrophages, many of which contain abundant intracytoplasmic droplets of cholesterol (foam cells). These macrophages are derived from monocytes which crossed the endothelium from the arterial lumen. They are not inert or end stage cells, but are highly activated, producing procoagulant tissue factor and a host of inflammatory cell mediators such as tumour necrosis factor α (TNF α), interleukins, and metalloproteinases. The connective tissue capsule which surrounds this inflammatory mass is predominantly collagen synthesised by smooth muscle cells. The portion of the capsule separating the core from the arterial lumen itself is the plaque cap.

The established stable plaque. In this cross section of a human coronary artery there is an established fibrolipid plaque with a core of lipid. The lipid core is separated from the lumen by the plaque cap. The plaque only occupies part of the circumference of the artery, leaving a segment of normal arterial wall.
The early stages of plaque development (AHA types I–III) are not associated with evidence of structural damage to the endothelium. Once plaque formation has progressed to stage IV, however, structural changes in the endothelium become almost universal.2 The endothelium over and between plaques shows enhanced replication compared to normal arteries, implying a degree of endothelial cell immaturity and abnormal physiological function. Focal areas of endothelial denudation occur over the plaque, exposing the underlying connective tissue matrix and allowing a monolayer of platelets to adhere at the site. Such ultramicroscopic thrombi are far too small to be visible on angiography or to impede flow, but may contribute to plaque smooth muscle cell growth by release of platelet derived growth factor.
Mechanisms of thrombosis
Thrombosis over plaques occurs because of two somewhat different processes. One is caused by an extension of the process of endothelial denudation so that large areas of the surface of the subendothelial connective tissue of the plaque are exposed. Thrombus forms which is adherent to the plaque surface (fig 2). This process has become known as endothelial erosion. Observational studies have linked endothelial cell loss to the proximity of macrophages. These macrophages are highly activated and cause endothelial cell death by apoptosis, and also by the production of proteases which cut loose the endothelial cells from their adhesion to the vessel wall.

Thrombosis caused by erosion. This human coronary artery is largely occluded by a mass of thrombus which is adherent to the surface of a plaque. The plaque itself is intact.
The second mechanism for thrombus formation is plaque disruption (synonyms rupture, fissuring) (fig 3). Here the plaque cap tears to expose the lipid core to blood in the arterial lumen. The core area is highly thrombogenic, containing tissue factor, fragments of collagen, and crystalline surfaces to accelerate coagulation. Thrombus forms initially in the plaque itself which is expanded and distorted from within; thrombus may then extend into the arterial lumen (fig4).

Thrombosis caused by disruption. The cap of a plaque has torn and projects up into the lumen. Thrombus has formed within the original lipid core from where it projects into, but does not totally occlude, the lumen. This is the typical lesion of unstable angina.

Thrombosis caused by disruption. The cap of the plaque has torn and thrombus within the lipid core extends into and occludes the lumen. This is the typical lesion of acute myocardial infarction.
Plaque disruption, like endothelial erosion, is a reflection of enhanced inflammatory activity within the plaque.3 The cap is a dynamic structure within which the connective tissue matrix, upon which its tensile strength depends, is constantly being replaced and maintained by the smooth muscle cell. The inflammatory process both reduces collagen synthesis by inhibiting the smooth muscle cell and causes its death by apoptosis. Macrophages also produce a wide range of metalloproteinases capable of degrading all the components of the connective tissue matrix, including collagen. These metalloproteinases are secreted into the tissues in an inactive form and then activated by plasmin. Metalloproteinase production by macrophages is upregulated by inflammatory cytokines such as TNFα. Plaque disruption is therefore now seen as an auto-destruct phenomenon associated with an enhanced inflammatory activation.
The relative importance of disruption and erosion as triggers of thrombosis may vary between different patient groups. Disruption is the predominant cause (> 85%) of major coronary thrombi in white males with high plasma concentrations of low density lipoprotein (LDL), and low concentrations of high density lipoprotein (HDL). In contrast, in women endothelial erosion is responsible for around 50% of major thrombi.4-6 The distinction between erosion and disruption is not necessarily of major clinical importance. Both processes depend on enhanced inflammatory activity within the plaque and appear equally responsive to lipid lowering. Disruption has an intraplaque component more resistant to fibrinolytic treatment, while in erosion the thrombus is more accessible. This potential advantage is, however, offset by erosion related thrombi tending to occur at sites where the pre-existing stenosis was more severe. In women there is also a form of thrombosis caused by endothelial erosion over plaques which do not contain lipid or have a major inflammatory component.6 This type of disease is rare and arguably distinct from conventional atherosclerosis, and may be smoking related.
The vulnerable plaque concept
Analysis of plaques which have undergone disruption has been used to determine characteristics which may indicate currently stable plaques whose structure and cell content makes them likely to undergo an episode of thrombosis in the future (vulnerable plaques).
There is widespread unanimity7 in the belief that these features are:
a large lipid core occupying at least 50% of the overall plaque volume
a high density of macrophages
a low density of smooth muscle cells in the cap
a high tissue factor content
a thin plaque cap in which the collagen structure is disorganised.
All of these markers of plaques at future risk are likely to be the direct result of macrophage activity, which enlarges the core and thins the cap.
The risk of any subject with coronary artery disease having a future acute event will depend on the number of these vulnerable plaques which are present rather than on the total number of plaques. Patients, however, vary in the number of vulnerable plaques which are present in the coronary arteries—this variation explains why one individual has a series of infarcts at regular intervals while another individual has an infarct without further events for 10 or even 20 years.
The sequence of thrombotic events
The thrombi which occur either in disruption or erosion circumstances are dynamic and evolve in stages. In disruption the initial stage occurs within the lipid core itself and is predominantly formed of platelets. As thrombus begins to protrude into the lumen the fibrin component increases, but any surface exposed to the blood in the lumen will be covered by activated platelets. While antegrade flow continues over this exposed thrombus, clumps of activated platelets are swept down into the distal intramyocardial arteries as microemboli (fig5). Thrombus may grow to occlude the artery, leading to a final stage in which there is a loose network of fibrin containing large numbers of entrapped red cells. This third and final stage thrombus may propagate distally after the onset of myocardial infarction. The final stage of occlusive thrombus has a structure making it very susceptible to either natural or therapeutic lysis, but this will expose the deeper and earlier thrombus which is more resistant to lysis.

Platelet embolisation. Any thrombus (red) which protrudes into the arterial lumen but does not occlude has the surface covered by a layer of activated platelets strongly expressing the IIb/IIIa receptor. Clumps of these platelets are swept down into the myocardium vascular bed.
Symptomology in relation to coronary thrombi
Episodes of plaque disruption which are almost entirely associated with intraplaque thrombus are associated with the onset or exacerbation of stable angina caused by a sudden increase in plaque volume.
Thrombi which project into but do not occlude the lumen (mural thrombi) are the basis of unstable angina. The intermittent attacks of myocardial ischaemia at rest are caused by several potential mechanisms.
The thrombus may intermittently wax and wane in size and become occlusive for relatively short periods of time.
There may be intense local vasoconstriction. Many disrupted plaques are eccentric, with the retention of an arc of normal vessel wall in which constriction can reduce blood flow.
Platelet deposition is a known potent stimulus for local smooth muscle constriction.
Embolisation of platelet aggregates into the intramyocardial vascular bed both block smaller arteries in the size range of 50−100 μm external diameter and cause vasoconstriction within the myocardium. Necropsy studies show a strong correlation between such platelet thrombi and microscopic foci of myocyte necrosis.8 ,9
Clinical correlations
Much of the work described so far is based on necropsy observations but these have been extended and amplified by observations made in life to give the dynamic dimension.
Acute myocardial infarction
It is difficult now to perceive why coronary thrombosis was regarded 25 years ago as an inconstant and irrelevant consequence of acute infarction rather than its prime cause. Once angiography was carried out soon after the onset of infarction, and it was realised that the subtending artery was totally blocked but spontaneously reopened with time in many cases (and that this reopening was accelerated by fibrinolytic treatment), thrombosis was seen as a major causal factor in occlusion. Suddenly the clinical world found thrombi to be both dynamic and important. Pathologists had thought thrombi were important but did not realise how dynamic they could be. Sequential angiograms taken over some years in patients with chronic ischaemic heart disease also changed perceptions. It was realised that a significant proportion of the thrombotic occlusions causing infarction did not develop at sites where there was pre-existing high grade stenosis, or even a plaque identified at all. Sixty eight per cent of the occlusions leading to acute infarction were judged to have caused less than 50% diameter stenosis previously, while only 14% developed on high grade stenoses of more than 70% diameter in a recent review of the literature.10
The advent of intravascular ultrasound has confirmed that many stable coronary plaques are angiographically invisible because of arterial remodelling. In this process, described so well by Glagov,11 the artery is seen to respond to plaque growth by increasing its cross sectional area while retaining normal lumen dimensions. Angiography cannot and does not predict the sites and risk of future infarction. It is true that chronic high grade stenoses do progress to occlude, but this is a slow process and is often caused by erosion type thrombosis, and is not associated with acute infarction due to collateral flow. For example, 24% of lesions occluding more than 80% by diameter will progress to chronic total occlusion by five years.
The magnitude of episodes of disruption varies widely. At one extreme the plaque has a crack or fissure only, and the large thrombotic response appears out of proportion to the stimulus. Such events are easily treated by lysis to give a lumen size which is little different from the previous state or event taken to be a normal artery. At the other extreme a plaque undergoes complete disintegration, occluding the lumen with a mixture of plaque content and thrombus. Another form is where the artery is occluded by the thrombus expanding the plaque from within. These more complex types of disruption occlusion will be more likely to respond to primary angioplasty. The exact morphology of disrupted plaques causing occlusion cannot, however, be determined in vivo by any current methodology.
Transmural regional acute myocardial infarction is caused by a coronary artery occlusion which develops over a relatively short time frame of a few hours and persists for at least 6–8 hours. The infarcted tissue is structurally suggestive of a homogenous entity—that is, all the myocardium involved died at around the same time. Non-transmural regional infarcts (non-Q wave) have a different structure which is built up by the coalescence of many small areas of necrosis of very different ages. This pattern of necrosis characteristically follows crescendo unstable angina and appears to be caused by repetitive episodes of short lived occlusion or platelet embolisation, or both. A further factor in limiting the spread of necrosis and preserving the subpericardial zone is the existence of prior collateral flow in the affected artery.
Unstable angina
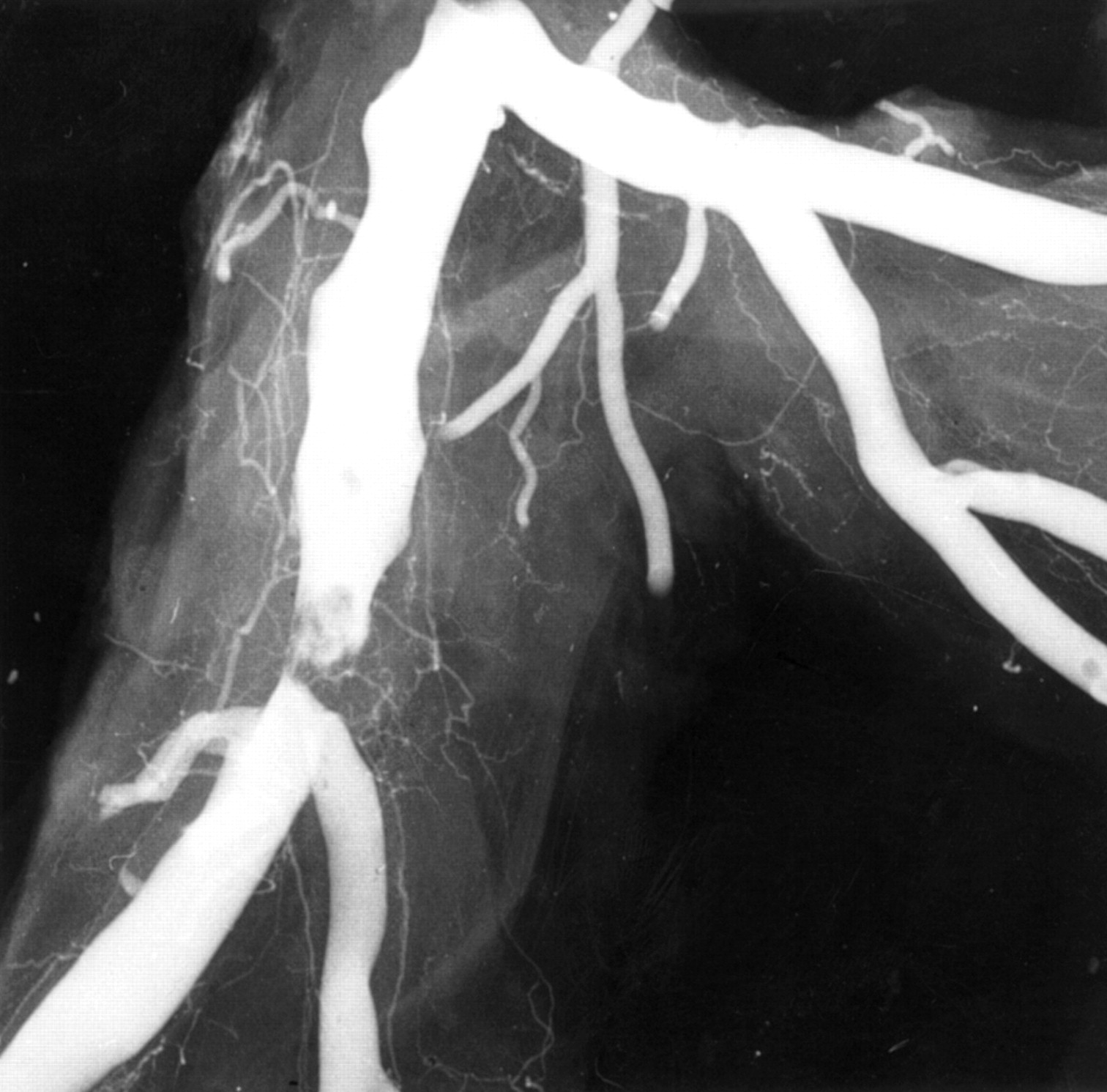
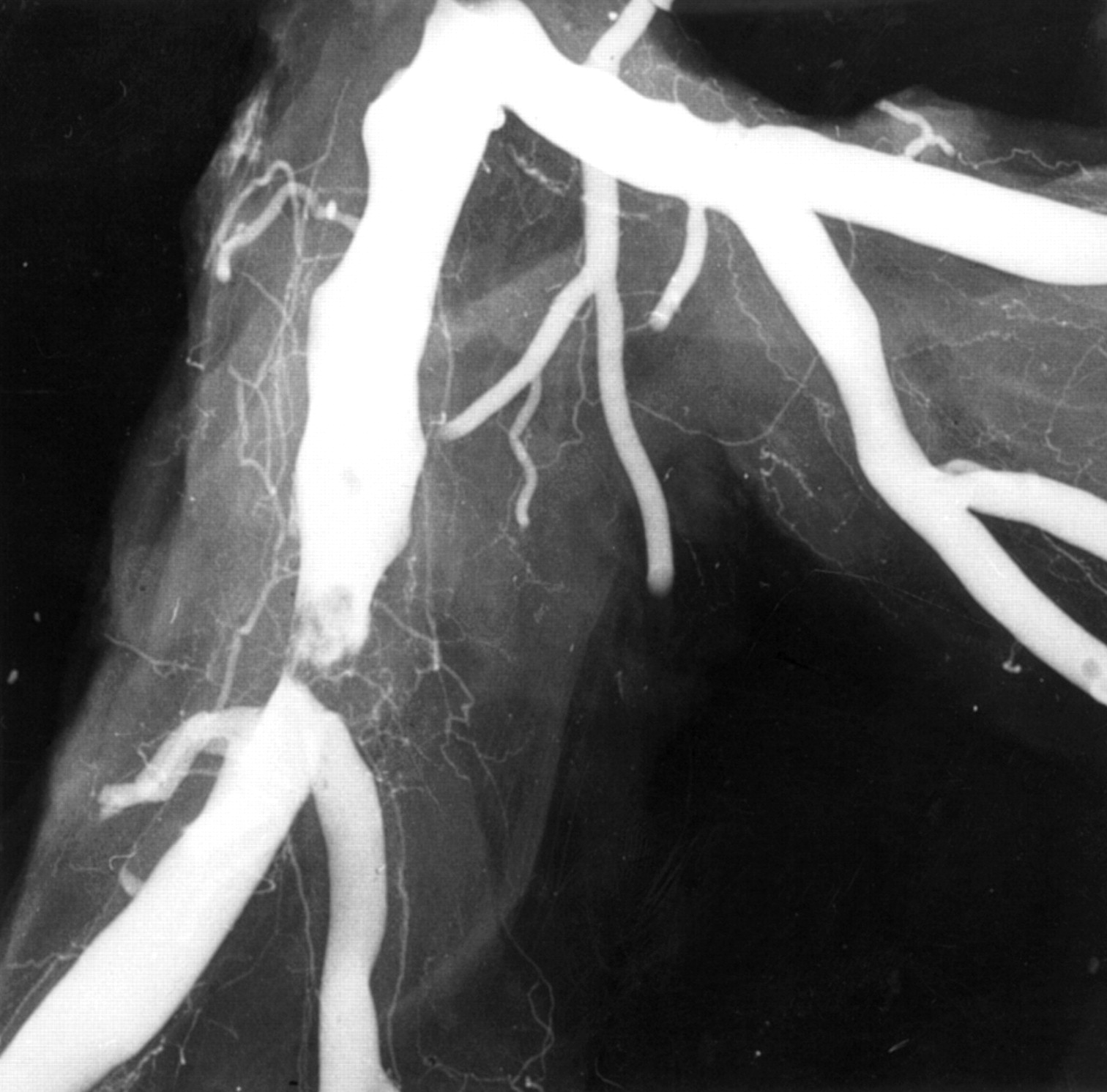
The challenge of understanding the pathophysiology of unstable angina is the wide spectrum of clinical severity.12Necropsy studies are inevitably biased toward the worst outcome, but within this limitation show unstable angina to be caused by disrupted plaques with exposed mural thrombus and retention of antegrade flow in the artery. This feature of some persistent antegrade flow is all that separates the vascular lesion of unstable angina from that of acute infarction. The persistence of the thrombotic process so that it neither progresses to occlude nor resolves to heal represents a balance between prothrombotic and antithrombotic factors. Confirmation of plaque disruption and thrombosis as the basis for severe unstable angina has come from angiography in vivo where type II lesions with irregular overhanging edges and intraluminal filling defects (fig 6) representing thrombus are found.13 These angiographic appearances are rare in stable angina. Type II lesions have been shown to be disrupted plaques by pathology studies. Angioscopy has directly observed torn plaque caps in vivo and intravascular ultrasound has also identified disrupted plaques in vivo. Atherectomy studies comparing tissue from plaques thought to be responsible for stable and unstable angina have shown very consistent results. A significant proportion, but not all, of samples from unstable angina contain thrombus, while most samples from stable angina, but not all, do not contain thrombus. The absence of thrombus in unstable angina is in part related to the time delay between acute symptoms and atherectomy.14
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Angiogram of plaque disruption. In this postmortem angiogram there is a typical type II eccentric ragged stenosis with an overlying intraluminal filling defect indicating thrombus over the plaque.
Samples taken some weeks after the last episode of rest pain often show accelerated smooth muscle proliferation—that is, the healing process rather than the acute thrombotic process. The presence of thrombus in plaque causing stable angina highlights the role of subclinical disruption or erosion in plaque growth. The pathological changes in plaques causing unstable angina expose thrombus in an artery in which antegrade flow continues. Platelet emboli into the myocardium cause microscopic foci of necrosis which are the basis of the increased concentrations of troponin T found in the blood in a proportion of cases of unstable angina.
The problem of the pathophysiology of unstable angina lies in patients who have milder and persistent rest pain over months or even years. The perception is that these cases are related to vasomotor tonal abnormalities often occurring at specific sites in the coronary artery tree. Why one such plaque should lead to local spasm is unclear—one suggestion is that there is local endothelial damage and repetitive ultramicroscopic thrombosis. The preponderance of literature reports of such vasospastic angina from Japan suggests there may be racial or geographic differences in the pathogenesis of this form of unstable angina.
Plaque disruption: the healing process
The great majority of episodes of plaque disruption do not cause a major event such as infarction or death. Minor episodes of erosion or disruption are often clinically silent but do contribute to the episodic progression of coronary artery disease seen on sequential angiography.
Thrombus will be removed by natural lysis to some extent and is also associated with “passification”, a term implying that the exposed collagen becomes less active in causing platelet adhesion probably due to being coated by natural heparinoids. Any residual thrombus which is still present after 36 hours will provoke smooth muscle cell migration into the area, with the production of new connective tissue which smooths out the surface and restores plaque integrity. The final result will be a stable lesion which may cause anything from chronic total occlusion to only a minor increase from the pre-existing degree of stenosis. The process, however, takes weeks and residual thrombi in the base of the exposed lipid core act as a nidus for a further thrombotic event at the same site for up to six months.
The risk of progressing to complete occlusion on angiography within a year of an episode of unstable angina, or after lysis for acute myocardial infarction, is far higher if the culprit lesion has an irregular outline on the initial angiogram taken immediately after the acute event.15
Mechanisms of myocardial ischaemia in non-occluding coronary thrombosis
Distal embolisation of aggregates of platelets
Intermittent total occlusion
Spasm at thrombus site
Inflammation and coronary atherosclerosis
Atherosclerotic plaques are the site of an inflammatory reaction which is of equal intensity to that found in the synovium in acute rheumatoid arthritis. The volume of any individual plaque in the coronary arteries is small, but most individuals have many plaques in the carotid artery and aorta which can be up to 2 cm in length. It is therefore not surprising that there may be elevation in systemic markers of inflammation such as fibrinogen and C reactive protein in subjects with chronic coronary atherosclerosis. The physicians health study shows that the difference between the lowest and highest quartiles of C reactive protein concentration is a threefold increase in the risk of a future acute event.16 The actual concentrations of C reactive protein are not, however, high and such estimates give assessment of group rather than individual risk. One explanation is that the subjects with the highest concentrations of C reactive protein have the largest plaque mass. The link between systemic markers of chronic inflammation and acute coronary events may, however, be more complex. There is experimental evidence that upregulation of systemic inflammation will have a secondary affect of enhancing inflammatory activity in the plaque.17 ,18 On this basis any factor which increases systemic inflammation would potentially trigger plaque instability and increase the risk of unstable angina. Causes of such systemic low grade inflammation include infection by chlamydia or helicobacter, diseases such as rheumatoid arthritis, and chronic dental sepsis.
Lipid lowering, infection, and acute coronary events
The current view of atherosclerosis is that the prime stimulus for plaque inflammation is the reaction between oxidised LDL and the macrophage. Significant reduction of plasma lipids in animal models of atherosclerosis has a profound effect on the plaque morphology, with a reduction in both macrophage numbers and activation products including metalloproteinases. The resultant plaque will be less inflammatory, and smooth muscle cell numbers and the collagen content rise. Although not reduced greatly in size, the plaque would be at far less risk of thrombosis. Similar changes in human plaques would explain the consistent benefit observed in a reduction of acute events in all the statin based trials.
Plaques at risk of future thrombotic events are characterised by:
Large lipid cores (> 50% overall plaque volume)
Thin caps
High densities of macrophages and high levels of expression of tissue factor and metalloproteinases
Low densities of smooth muscle cells
Even allowing for poor patient compliance, the need for at least 18 months for the benefit to appear, and inadequate lipid lowering, acute ischaemic events still occur in treated patients. This suggests there may be non-lipid dependent factors enhancing plaque inflammation. One such factor is direct invasion of the plaque by chlamydia. These organisms exist within macrophages and potentially could upregulate the production of inflammatory mediators within the plaque.18Therapeutic trials of antichlamydial drugs to reduce acute coronary event rates are currently underway but results so far are contradictory. It seems likely that there are many other factors which enhance the inflammatory activity of the plaque, but the primary stimulus remains the conversion of plasma LDL in the intima to a proinflammatory product.19 In keeping with this view is the consistent message from animal models of atherosclerosis that reducing plasma lipid concentrations strikingly reduces all the inflammatory processes in the plaque.20