Article Text

Statistics from Altmetric.com
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a myocardial disease, often familial, that is characterised pathologically by fibrofatty replacement of the right ventricular myocardium, and clinically by ventricular arrhythmias of right ventricular origin which may lead to sudden death, mostly in young people and athletes.1-5 The term “dysplasia” was originally used to describe an entity that was considered to be the result of a developmental defect of the right ventricular myocardium.1 A better understanding of clinical manifestations as well as morphologic findings does not support the theory of a congenital absence of the myocardium, but is in keeping with a non-ischaemic, ongoing atrophy of the right ventricular myocardium, most likely genetically determined, which becomes symptomatic in adolescents and young adults.2-4 On the basis of its nature of progressive heart muscle disease of unknown aetiology, ARVC has been more appropriately included among the cardiomyopathies in the recent classification proposed by the task force of the World Health Organization/International Society and Federation of Cardiology. Although several theories have been advanced, the aetiopathogenesis of ARVC is still unknown.5
Pathologic features
The most striking pathologic feature of ARVC is the diffuse or segmental loss of the myocardium of the right ventricular free wall and its replacement by fibrofatty tissue (fig 1); it is frequently transmural and accounts for aneurysmal dilations of the diaphragmatic, apical, and infundibular regions (so called “triangle of dysplasia”) in nearly 50% of the cases in the necropsy series.2-4 A wave front progression of the pathological process occurs from the subepicardium to the endocardium, so that residual myocardium is confined to the inner subendocardial layer and to the trabeculae of the right ventricle, where islands of surviving myocardial cells are scattered throughout the fibrofatty tissue. Patchy acute myocarditis with myocyte death and round cell (mostly T lymphocytes) inflammatory infiltrates are present in nearly two thirds of the cases.
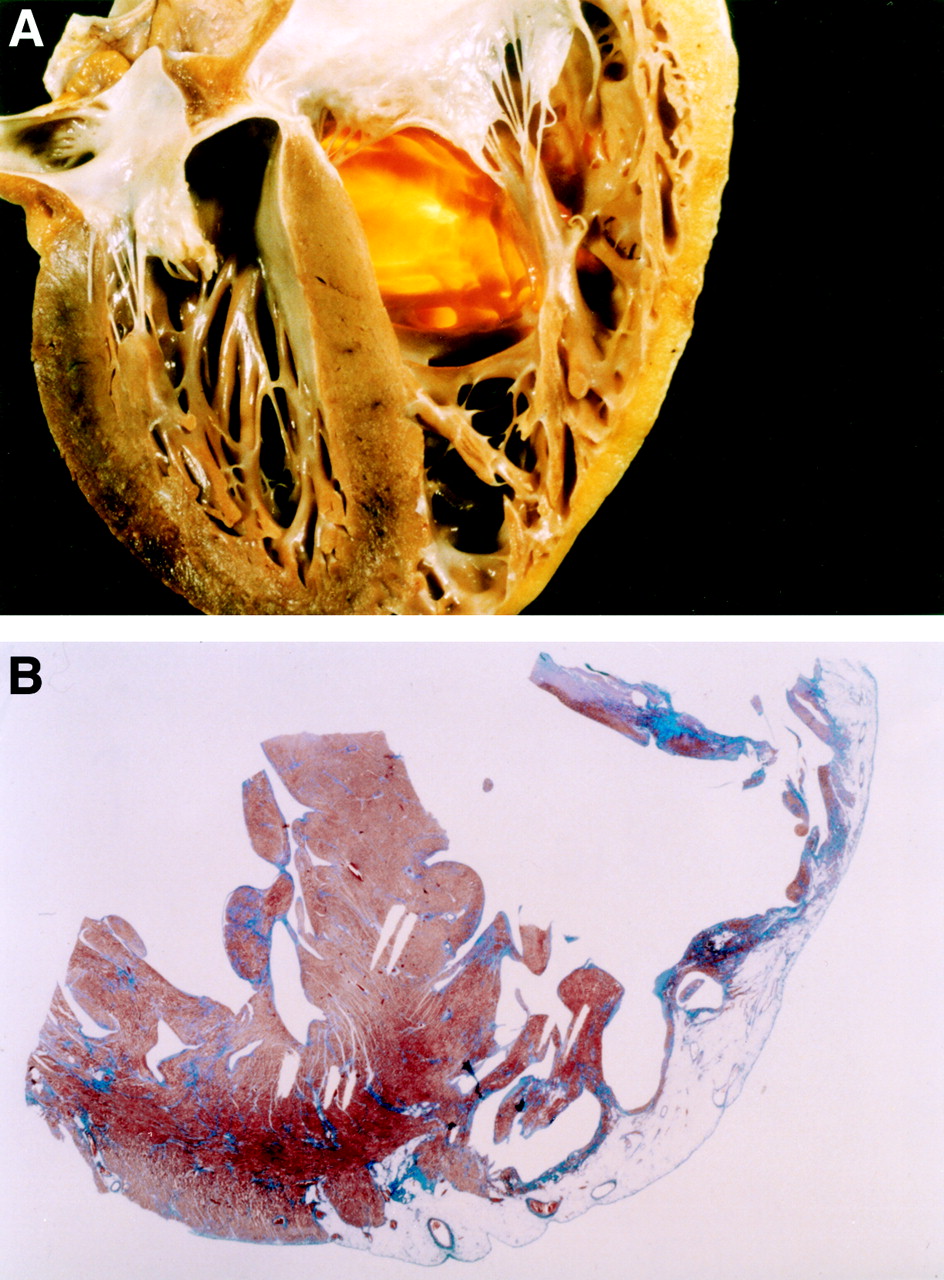
Morphologic features in a 25 year old man who died suddenly from arrhythmogenic right ventricular cardiomyopathy. (A) Four chamber view cut of the heart specimen showing the transmural fatty replacement of the right ventricular free wall and the translucent infundibulum. (B) Panoramic histologic view of the same heart confirming that the myocardial atrophy is confined to the right ventricle and substantially spares the interventricular septum as well as the left ventricular free wall (trichrome Heidenhain × 3). Reproduced from Basso C, Corrado D, Rossi L, et al. Morbid anatomy. In: Nava A, Rossi L, Thiene G, eds. Arrhythmogenic right ventricular cardiomyopathy—dysplasia. Elsevier, Amsterdam 1997, pp 71-86, with permission of the publisher.
Two morphological variants of ARVC have been reported.2The fatty form is exclusively confined to the right ventricle and involves predominantly the apical and infundibular regions. It is characterised by partial or almost complete substitution of the myocardium by fatty tissue without wall thinning (4–5 mm). There is evidence of myocardial degeneration and death in about half of the cases, in the absence of significant fibrous tissue and inflammatory infiltrates. The left ventricle and the interventricular septum are typically spared. In thefibrofatty variant the adipose infiltration is associated with significant replacement type fibrosis, thinning of the right ventricular wall (< 3 mm) (fig 1A), aneurysmal dilatation, and inflammatory infiltrates. There is usually involvement of the diaphragmatic wall underneath the posterior leaflet of the tricuspid valve; the left ventricle and, more rarely, the ventricular septum may be involved to a lesser extent.
The replacement of the right ventricular myocardium by fibrofatty tissue has been related to three basic mechanisms3:
Apoptosis or programmed cell death;
Inflammatory heart disease with a spectrum of clinical presentations ranging from acute myocarditis to fibrous healing, which in severe forms may involve both right and left ventricles and may lead to congestive heart failure mimicking dilated cardiomyopathy;
Myocardial dystrophy which might reflect a genetically determined atrophy.
In this setting, a genetic propensity to infectious and/or immune reaction may explain the occurrence of myocarditis.
Clinical diagnosis
The most common clinical manifestations of ARVC consists of ventricular arrhythmias with left bundle branch block (LBBB) morphology, ECG depolarisation/repolarisation changes mostly localised to right precordial leads, and global and/or regional dysfunction and structural alterations of the right ventricle.1-6However, patients with a clinical diagnosis of ARVC based on typical findings, such as right precordial ECG changes, right ventricular arrhythmias, and structural and functional right ventricular abnormalities, represent only one extreme of the disease spectrum. A number of cases are not recognised because they are asymptomatic until the first presentation with cardiac arrest or are difficult to diagnose through conventional non-invasive methods. In this regard, a prospective investigation on sudden death in the young in the Veneto region of Italy showed that nearly 20% of fatal events in young people and athletes were caused by concealed ARVC. At the other extreme of the spectrum are patients in whom the diagnosis of ARVC was not recognised at the onset of their symptoms, who present in later years with congestive heart failure with or without ventricular arrhythmias, and are often misdiagnosed as having dilated cardiomyopathy.4
Standardised diagnostic criteria have been proposed by the study group on ARVC of the working group on myocardial and pericardial disease of the European Society of Cardiology and of the scientific council on cardiomyopathies of the International Society and Federation of Cardiology.6 This task force was established because it was realised that the diagnosis of ARVC may be difficult because of several problems with the specificity of the ECG abnormalities, the different potential aetiologies of ventricular arrhythmias with an LBBB morphology, with the assessment of the right ventricular structure and function, and with the interpretation of endomyocardial biopsy findings. According to the task force guidelines, the diagnosis of ARVC is based on the presence of major and minor criteria encompassing genetic, electrocardiographic, arrhythmic, morphofunctional, and histopathologic factors (table 1). Based on this classification the diagnosis of ARVC would be fulfilled in the presence of two major criteria or one major plus two minor or four minor criteria from different groups. Although these guidelines represent a useful clinical approach to ARVC diagnosis, optimal assessment of diagnostic criteria requires a prospective evaluation from a large patient population.
Criteria for diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC)6
Genetics
A familial background have been demonstrated in nearly 50% of ARVC cases, with an autosomal dominant pattern of inheritance.5 ,7 The involved genes and the molecular defects causing the disease are still unknown. However, seven ARVC loci have been identified so far, two of which are in close proximity of chromosome 14 (14q23-q24 and 14q12-q22),8 and the others on chromosome 1 (1q42-q43), chromosome 2 (2q32.1-q32.2), chromosome 3 (3p23), and chromosome 10 (p12-p14). An autosomal recessive variant of ARVC that is associated with palmoplantar keratosis and woolly hair (so called “Naxos disease”) has been mapped on chromosome 17. Genes encoding for actinin and keratin have been considered as potential candidates for the dominant and recessive variant of ARVC, respectively. It is noteworthy that in the Padua experience about 50% of the ARVC families undergoing clinical and genetic screening did not show linkage with any of the known chromosomal loci. Therefore, further genetic heterogeneity can be postulated. Although a preclinical diagnosis of ARVC by DNA characterisation is warranted, at the present time a genetic test for screening is not currently available.
Depolarisation/repolarisation abnormalities
ECG abnormalites are detected in up to 90% of ARVC patients.5 The most common abnormality consists of T wave inversion in the precordial leads exploring the right ventricle (V1–V3) (fig 2). Inversion of T waves is often associated with a slight ST segment elevation (< 0.1 mV). These repolarisation changes are not specific and are considered only minor diagnostic criteria because they may be a normal variant in females and in children aged less than 12 years, or may be secondary to a right bundle branch block, either isolated or in the setting of a congenital heart disease accounting for a right ventricular overload.

Twelve lead ECG obtained at preparticipation screening in a 19 year old football player who subsequently died from ARVC during a competitive game. Note the typical abnormalities consisting of inverted T waves from V1 to V4 and isolated premature ventricular beats with an LBBB morphology.
The wide spectrum of ECG abnormalities reflecting a delayed right ventricular activation includes complete or incomplete right bundle branch block, prolongation of right precordial QRS duration, and postexcitation epsilon waves—that is, small amplitude potentials occurring after the QRS complex at the beginning of the ST segment. Correlation between surface ECG and epicardial mapping has shown that these ECG changes reflect an intraventricular myocardial (“parietal block”) defect rather than a specialised conduction system (“septal block”) conduction defect.9 Both right precordial QRS prolongation and epsilon waves are deemed major diagnostic criteria. Localised prolongation of QRS complex in V1–V3 to more than 110 ms is a relatively sensitive and specific diagnostic marker; it mostly accounts for the QT dispersion across the 12 leads which has been reported to relate to the risk of sudden death. Epsilon waves are uncommon on standard 12 lead ECGs but can be detected in over 30% of ARVC patients in the form of late potentials by high resolution ECG and signal averaging techniques. Late potentials are fragmented low amplitude potentials in the terminal portion of the QRS complex. They reflect areas of slow intraventricular conduction which may predispose to re-entrant ventricular arrhythmias. The underlying substrate consists of islands of surviving myocardium interspersed with fatty and fibrous tissue, accounting for fragmentation of the electrical activation of the ventricular myocardium. In ARVC, late potentials are not specific for re-entrant ventricular arrhythmias and are better correlated with the extension of right ventricular involvement and with the disease progression over the time. Recently, a relation between late potentials, amount of replacement-type fibrous tissue, and degree of right ventricular dysfunction has been reported. Less common ECG abnormalities include P waves exceeding 2.5 mV in amplitude, low voltage QRS complex in peripheral leads, and T wave inversion in the inferior leads.
Ventricular arrhythmias
Although there are some asymptomatic ARVC patients who are recognised by chance or in the setting of a family screening, the most usual clinical presentation of the disease is as symptomatic ventricular arrhythmias of right ventricular origin, which characteristically occur during exercise. Related symptoms consist of palpitations, presyncope, and syncope. Ventricular arrhythmias range from isolated premature ventricular beats (fig 2) to sustained ventricular tachycardia (VT) with an LBBB morphology, or ventricular fibrillation (VF) leading to sudden cardiac arrest. The QRS morphology and the mean QRS axis during VT reflects its site of origin: an LBBB with inferior axis suggests the right ventricular outflow tract, while an LBBB with superior axis suggests the right ventricular inferior wall. It is not uncommon for patients with advanced ARVC to show several morphologies of VT, suggesting multiple right ventricular arrhythmogenic foci. VTs with LBBB pattern are not specific for ARVC.
In the presence of right ventricular tachycardias, the following structural heart disease characterised by right ventricular involvement should be ruled out before considering the diagnosis of ARVC: congenital heart disease, such as repaired tetralogy of Fallot, Ebstein anomaly, atrial septal defect, and partial anomalous venous return; acquired disease such as tricuspid valve disease, pulmonary hypertension, and right ventricular infarction; and bundle branch re-entry complicating a dilated cardiomyopathy. Once underlying structural right ventricular disease is excluded, the differential diagnosis should include a Mahaim pre-excitated atrioventricular re-entry tachycardia or an idiopathic right ventricular outflow tract tachycardia. It is often difficult to differentiate ARVC from the latter condition, which is usually benign and non-familial. It is still debated whether right ventricular outflow tract tachycardia represents a minor form of ARVC, as suggested by the right ventricular structural abnormalities often detected by magnetic resonance imaging (MRI).
The true incidence of VF leading to sudden cardiac arrest in patients with ARVC remains unknown because many cases are discovered only at post mortem. VF is relatively rare in patients with known ARVC undergoing medical treatment of symptomatic ventricular tachycardia, although some cases of rapid, haemodynamically unstable or prolonged VT may degenerate into VF. On the other hand, abrupt VF is the most likely mechanism of instantaneous sudden death in previously asymptomatic young people and athletes with concealed ARVC.10 Whether VF in this subset of patients is related to an acute phase of disease progression, either because of myocyte necrosis–apoptosis or inflammation, remains to be established.
Imaging of right ventricular morphofunctional abnormalities
Demonstration of right ventricular morphofunctional abnormalities by echocardiography, angiography, and MRI is a major criterion for diagnosing ARVC.5 Functional and structural abnormalities consist of global right ventricular dilatation with or without ejection fraction reduction and left ventricular involvement; segmental right ventricular dilatation with or without dyskinesia (aneurysms and bulgings); and wall motion abnormalities such as ipo-akinesia or dyskinesia.
All the imaging techniques are associated with significant limitations in the diagnostic accuracy for detecting right ventricular changes. Right ventricular angiography is usually regarded as the gold standard for the diagnosis of ARVC. Angiographic evidence of akinetic or dyskinetic bulgings localised in infundibular, apical, and subtricuspidal regions has a high diagnostic specificity (over 90%).11 Large areas of dilatation akinesia with an irregular and “mamillated” aspect, most often involving the inferior right ventricular wall, are also significantly associated with the diagnosis of ARVC. However, considerable interobserver variability regarding the visual assessment of right ventricular wall motion abnormalities by contrast angiography have been reported.
Compared with right ventricular angiography, echocardiography is a non-invasive and widely used technique, and represents the first line imaging approach in evaluating patients with suspected ARVC or in screening family members. Echocardiography also allows serial examinations aimed to assess the disease progression during the follow up of affected patients. Furthermore, echocardiography is a reliable technique for differential diagnosis of ARVC by easily ruling out other right ventricular diseases such as Ebstein anomaly, atrial septal defect, etc. Other than a visual assessment of wall motion and structural abnormalities, a quantitative echocardiographic evaluation of the right ventricle, including measurements of end diastolic cavity dimensions (inlet, outlet, and mean ventricular body), wall thickness, volume, and function, is mandatory in order to enhance the diagnostic accuracy. In the presence of the typical echocardiographic features, contrast angiography or MRI may be avoided, whereas borderline or apparently normal findings in patients with suspected disease requires further examination.
MRI is an attractive imaging method because it is non-invasive and has the unique ability to characterise tissue, specifically by differentiating fat from muscle.12 Recent studies have shown several limitations and a high degree of interobserver variability in the MRI assessment of free wall thinning and fatty deposition that are the most characteristic structural changes (fig 3). The right ventricular free wall is only 4–5 mm thick and the motion artifacts often result in insufficient quality/spectral resolution to quantify right ventricular wall thickness accurately. The normal presence of epicardial and pericardial fat also makes identification of true intramyocardial fat difficult. Some areas—such as the subtricuspidal region—are not easily distinguished from the atrioventricular sulcus which is rich in fat. There has been recent emphasis on functional methods such as right ventricular volume estimation with cine MRI. This approach also permits accurate assessment of right ventricular wall motion abnormalities and focal areas of dilatation with or without dyskinesia. In conclusion, although MRI is a promising technique for delineating right ventricular anatomy and function, as well as for characterising the composition of the right ventricular wall, its diagnostic sensitivity and specificity still need to be defined since the quality of images detected are, at the present time, largely subject to individual interpretation.

MRI findings in a 22 year old woman with a history of dizziness and sustained ventricular tachycardia with an LBBB pattern. Short axis view showing a dilated right ventricle with a brighter signal from a thin anterior free wall. Reproduced from Menghetti L, Basso C, Nava A, et al. Spin-echo nuclear magnetic resonance for tissue characterisation in arrhythmogenic right ventricular cardiomyopathy. Heart 1996;76:467–70, with permission of the publisher.
Radionuclide angiography is also an accurate non-invasive imaging technique for detection of global right ventricular dysfunction and regional wall motion abnormalities; its diagnostic concordance with right ventricular angiography is nearly 90%.
The diagnosis of ARVC at its early stages or in its concealed variants remains a clinical challenge by all imaging methods. Although these techniques appear to be accurate in detecting right ventricular structural and functional abnormalities in overt forms of ARVC, they are less sensitive in detecting subtle lesions.
Endomyocardial biopsy
A definitive diagnosis of ARVC relies on the histological demonstration of full thickness substitution of the right ventricular myocardium by fatty or fibrofatty tissue at postmortem examination. Transvenous endomyocardial biopsy has the potential for an “in vivo” demonstration of typical fibrofatty replacement of the right ventricular muscle and may increase the accuracy for the clinical diagnosis of ARVC, even though it has several diagnostic limitations. The sensitivity of endomyocardial biopsy is low owing to the segmental nature of the ARVC lesions and because the samples are usually taken from the septum for safety reasons, a region uncommonly involved by the disease. On the other hand, there is difficulty in differentiating ARVC from other causes of fatty infiltration of the right ventricular myocardium. In healthy subjects, particularly in the elderly, there is a normal amount of subepicardial adipose tissue which reflects the physiologic process of progressive involution of the right ventricle. Pathologic conditions which have been associated with fatty infiltration include chronic consummation of alcohol and inherited myopathies such as Duschenne/Backer muscular dystrophy. On the other hand, fibrosis can be observed in many cardiomyopathic and non-cardiomyopathic conditions. Histomorphometric criteria have been advanced in order to enhance the specificity of histopathologic diagnosis of ARVC at endomyocardial biopsy. A percentage of fat > 3% and of fibrous tissue > 40% with amounts of myocytes < 45% was considered a clear cut diagnostic border between ARVC and normal hearts or dilated cardiomyopathy.13 Although biopsy cannot be routinely recommended, it represents a histologic validation of clinical findings and may improve the diagnostic accuracy by excluding other cardiomyopathy or myocarditis conditions, both idiopathic and specific.
Prognosis
Natural history
The natural history of ARVC is predominantly related to the ventricular electrical instability which can precipitate arrhythmic cardiac arrest at any time during the disease course. Moreover, there is clinical and pathological evidence that ARVC is a progressive heart muscle disease. Long term follow up data from clinical studies indicate that the right ventricle may become more diffusely involved with time.14 Later in the natural history, the left ventricle may be progressively affected with subsequent biventricular failure. Recently a multicentred clinicopathologic investigation was carried out to define further the anatomoclinical profile of ARVC, with special reference to disease progression and left ventricular involvement.4 By examining 42 affected whole hearts, including those removed at transplant, and correlating pathologic findings with the patient's clinical history, the study demonstrated that at least in this subgroup, representing an extreme of the disease spectrum, ARVC can no longer be regarded as an isolated disease of the right ventricle. Macroscopic or histologic involvement of the left ventricle was found in 76% of hearts with ARVC; it was age dependent, more common in patients with longstanding clinical history, and it was progressive as evaluated by serial echocardiographic examinations. Moreover, left ventricular lesions were associated with clinical arrhythmic events, more severe cardiomegaly, inflammatory infiltrates, and heart failure.
At present, there is limited information about the clinical outcome of ARVC patients with overt disease and significant ventricular arrhythmias, and even less on asymptomatic affected family members. The following clinicopathologic phases can be considered5:
“Concealed” phase characterised by subtle right ventricular structural changes, with or without minor ventricular arrhythmias, during which sudden death may occasionally be the first manifestation of the disease, mostly in young people during competitive sports or intense physical exercise.
“Overt electrical disorder” in which symptomatic right ventricular arrhythmias possibly leading to sudden cardiac arrest are associated with overt right ventricular functional and structural abnormalities.
“Right ventricular failure” caused by the progression and extension of right ventricular muscle disease that provokes global right ventricular dysfunction with a relatively preserved left ventricular function.
Final stage of “biventricular pump failure” caused by pronounced left ventricular involvement. At this stage, ARVC mimics biventricular dilated cardiomyopathy of other causes leading to congestive heart failure and related complications such as atrial fibrillation and thromboembolic events.
Risk stratification
The main objective of management strategy is to prevent arrhythmic sudden death. However, there are no prospective and controlled studies assessing clinical markers which can predict the occurrence of life threatening ventricular arrhythmias. It has been established that sudden death may be the first manifestation of the disease, mostly in previously asymptomatic young subjects and athletes. Therefore all identified or suspected patients are at risk of sudden death even in the absence of symptoms or ventricular arrhythmias. The most challenging clinical dilemma is not whether to treat patients who already experienced malignant ventricular arrhythmias (secondary prevention), but to consider prophylactic treatment in patients with no or only minor symptoms in whom the disease has been diagnosed during family screening or by chance (primary prevention). Furthermore, ARVC is a progressive disease and the patient's risk of sudden death may increase with time.
The risk profile which emerges from retrospective analysis of clinical and pathologic series, including fatal cases, is characterised by young age, competitive sport activity, malignant familial background, extensive right ventricular disease with ejection fraction reduction and left ventricular involvement, syncope, and episodes of complex ventricular arrhythmias or VT.2-4 ,10 The baseline clinical study for assessment of the risk of sudden death consists of non-invasive routine clinical study including detailed clinical history (mostly addressing familial background and previous syncope), 12 lead ECG, 24 hour Holter monitoring, exercise stress testing, and signal averaged ECG. All affected patients, either symptomatic or asymptomatic, should undergo this first line evaluation, even if its positive predictive value for subsequent malignant ventricular arrhythmias remains to be established. Invasive, intracardiac electrophysiologic study with programmed ventricular stimulation should be reserved for patients symptomatic for sustained VT or VF, patients with syncopal episodes in whom non-invasive evaluation was negative, and asymptomatic patients with strongly positive findings such as history of premature sudden death, non-sustained ventricular tachycardia, and depressed right ventricular function. The major aims of electrophysiologic study are: (1) to assess the disease's arrhythmogenic potential by induction of VT or VF during the basic pacing protocol or after isoproterenol; (2) to evaluate haemodynamic consequences of sustained VT and its propensity to degenerate into VF; and (3) to establish the susceptibility of VT to be interrupted by antitachycardia stimulation, and its reinducibility in view of serial electropharmacologic studies or implantation of an automatic defibrillator. The value of electrophysiologic study in arrhythmic risk stratification of patients with ARVC remains to be validated by prospective investigations.
Treatment
Since the assessment of sudden death risk in patients with ARVC is still not well established, there are no precise guidelines to determine which are the patients who need to be treated and which is the best management approach.5 The therapeutic options include β blockers, antiarrhythmic drugs, catheter ablation, and implantable cardioverter defibrillator (ICD). At the present time, it is unclear how to best predict the efficacy of both pharmacological and non-pharmacological treatment in patients with ARVC; management of patients with ARVC is individualised and the strategies are based on the local experience of the different centres. Pharmacologic therapy is the first choice treatment of patients with well tolerated and non-life threatening ventricular arrhythmias. This subset of patients is usually treated empirically by β blockers and class I and III antiarrhythmic drugs. The evidence available suggests that either sotalol or amiodarone (alone or in combination with β blockers) are the most effective drugs with a relatively low proarrhythmic risk. The evaluation of efficacy of antiarrhythmic treatment may be based on symptom improvement, though a more reliable approach involves guiding treatment by serial 24 hour Holter monitoring and/or stress testing by demonstrating reduction in arrhythmic events.
In patients with sustained VT or VF, antiarrhythmic drug treatment guided by programmed ventricular stimulation with serial drug testing is the most effective therapeutic strategy, although its ability to prevent sudden death has not been proven. Non-pharmacological treatment is reserved for those patients with life threatening ventricular arrhythmias in whom drug treatment either is ineffective, is not applicable because of the inability to induce the clinical ventricular arrhythmias during electrophysiologic study, or is associated with serious side effects. Figure 4 is a flow chart outlining the management of ARVC patients with VT or VF. Patients with inducible VT during programmed ventricular stimulation undergo drug testing (mostly class I and III antiarrhythmic drugs) guided by serial electrophysiologic study. If a drug regimen can be found that prevents the induction of VT, the patient is discharged on the effective drug. Patients who remain inducible on different antiarrhythmic drugs are treated non-pharmacologically by an ICD (fig 5), except for rare cases with localised disease in whom catheter ablation of the VT may be an alternative option. Among patients with non-inducible VT, those who experienced spontaneous, haemodynamically well tolerated VT may be treated empirically by antiarrhythmic drug treatment, which is subsequently tested by serial 24 hour Holter monitoring and exercise stress testing. In case of no drug response, pace mapping directed catheter ablation may be attempted before implanting a prophylactic defibrillator. In patients with previous syncope or cardiac arrest and no inducible VT, the automatic cardioverter defibrillator represents the first option.

Treatment strategy in ARVC complicated by VT/VF.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
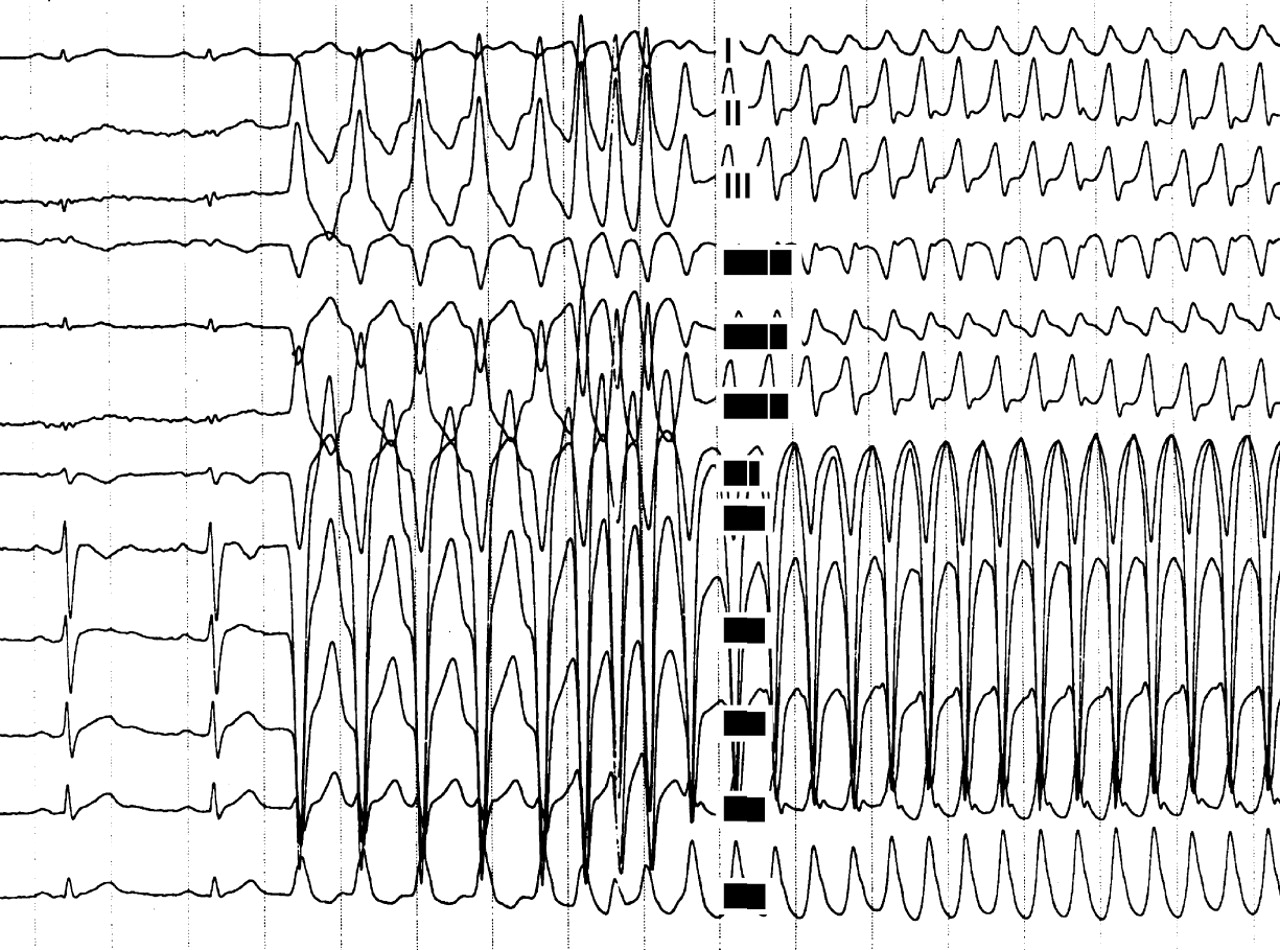
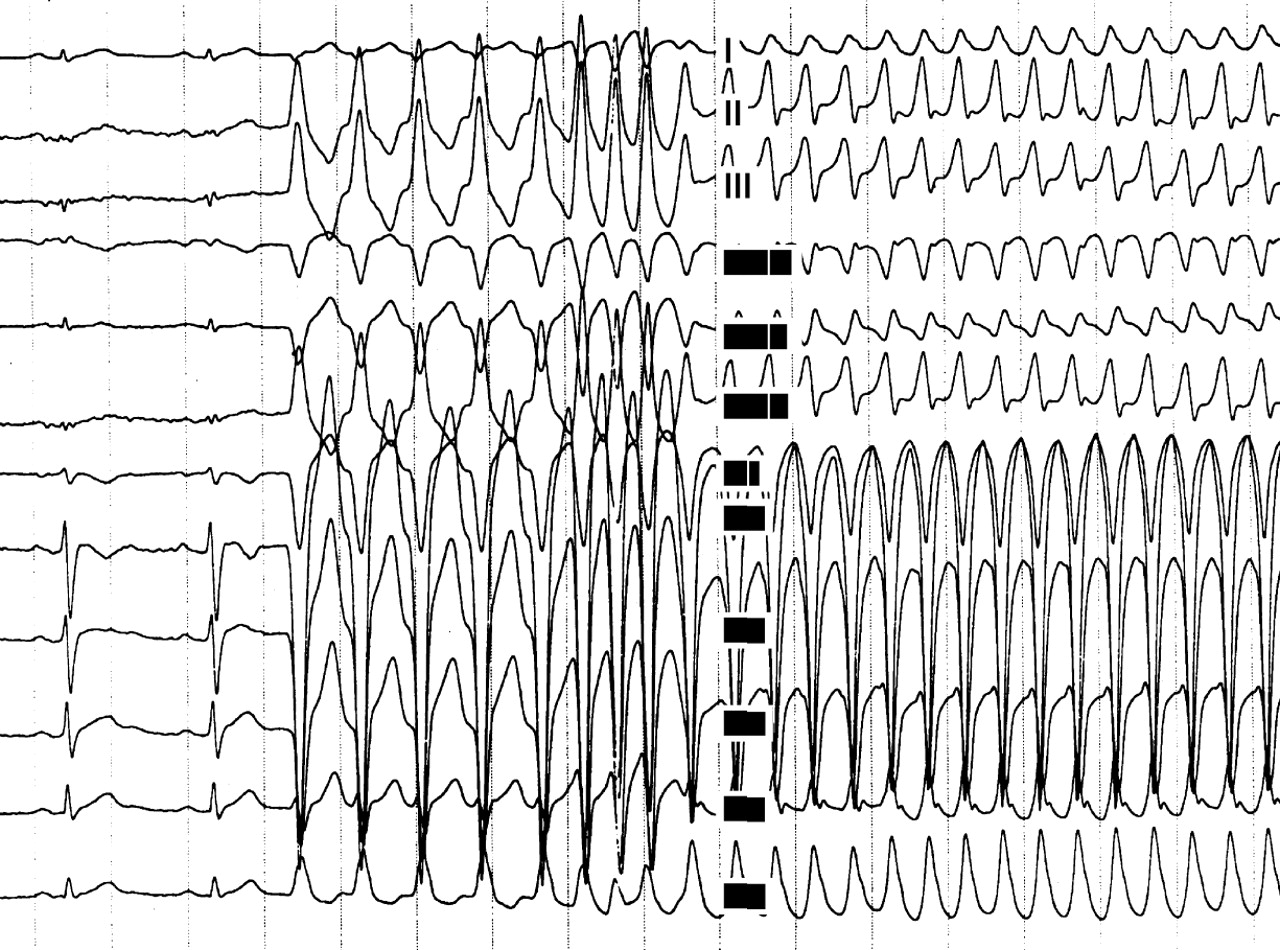
A 24 year old man affected by ARVC complicated by severe ventricular arrhythmias, with a recent family history of sudden death. (A) Twelve lead ECG during programmed ventricular stimulation: the first two beats at sinus rhythm show a low voltage QRS complex in the peripheral leads and T wave inversion in right precordial leads. After a drive of five paced ventricular beats, three extrastimuli induce a sustained ventricular tachycardia with an LBBB pattern and a cycle length of 250 ms, which was promptly interrupted by DC shock caused by the rapid haemodynamic deterioration. Serial antiarrhythmic drug testing, including sotalol, failed to identify any effective drug. (B) Chest radiography (60° left anterior oblique) of the same patient after implantation of a transvenous prophylactic automatic cardioverter defibrillator. Besides the atrial lead (a) and the double coil ventricular lead (b) for cardioversion, a third lead (c) was screwed onto the mid septum to assure a reliable sensing and pacing function. A B
Sotalol has been reported to be the most effective antiarrhythmic drug in the treatment of both inducible and non-inducible VT in ARVC, with overall efficacy rates of more than 68% and 82%, respectively.15 However, its efficacy in preventing sudden death remains to be established.
Catheter ablation
Although acute success rates of 60–90% have been reported with catheter ablation, VT recurrences are common (up to 60% of the cases) and may lead to sudden arrhythmic death. The progressive nature of the underlying disease which predisposes to the occurrence of new arrhythmogenic foci over time may explain this discrepancy between acute and long term follow up success rates. In this regard, recurring VTs usually show a QRS morphology other than that previously ablated. Therefore, catheter ablation should be reserved for particular clinical conditions such as drug refractory incessant VT or frequent recurrences of VT after defibrillator implantation. Haemodynamically stable and well tolerated VT, which is not inducible or is not suppressed by electrophysiologic study directed pharmacologic treatment, may represent a further indication, in the presence of very localised cardiomyopathic changes and a still preserved right ventricular function.
Implantable cardioverter defibrillator
The ICD is the most effective safeguard against arrhythmic sudden death. However, its precise role in changing the natural history of ARVC by preventing sudden and non-sudden death needs to be evaluated by a prospective study of a large series of patients. The implantable defibrillator is the treatment of choice in patients resuscitated from a cardiac arrest caused by rapid VT or VF. Other indications include: patients with symptomatic VT non-inducible at electrophysiologic study; patients in whom electrophysiologic study guided drug treatment is ineffective or is associated with severe side effects; patients with severe right ventricular involvement and poor tolerance of VT; and sudden death of a close family member. Although ICD may confer a survival benefit of up to 50% in patients with ARVC, there are potential complications associated with the device implantation which are related to the distinctive pathologic changes of the right ventricular wall. The very thin free wall predisposes to right ventricular perforation by the transvenous lead. The loss of the right ventricular myocardium with fibrofatty replacement underlies the difficulty in obtaining adequate R waves and pacing thresholds at the time of implantation (fig 5B). Moreover, undersensing or pacing exit block may occur during the follow up as a consequence of the progression of the myopathic process, leading to right ventricular myocardial disappearance. Finally, inappropriate treatment owing to sinus tachycardia and lead dislodgement has been described in more active young patients undergoing implantation of the device.
Heart failure
In patients in whom ARVC has progressed to severe right ventricular or biventricular systolic dysfunction with risk of thromboembolic complications, treatment consists of current therapy for heart failure, including diuretics, angiotensin converting enzyme inhibitors, and digitalis, as well as anticoagulants. In case of refractory congestive heart failure, the patients may become candidates for heart transplantation.